

This is a preprint.
Entropy tree networks of residue dynamics encode protein allostery
- PMID: 40501711
- PMCID: PMC12154606
- DOI: 10.1101/2025.05.28.656549
Entropy tree networks of residue dynamics encode protein allostery
Abstract
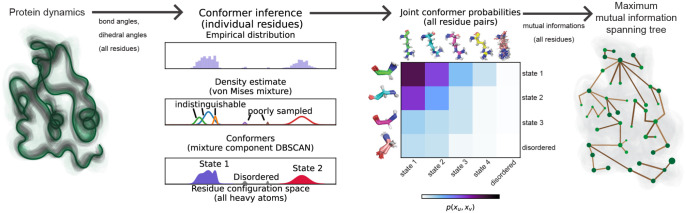
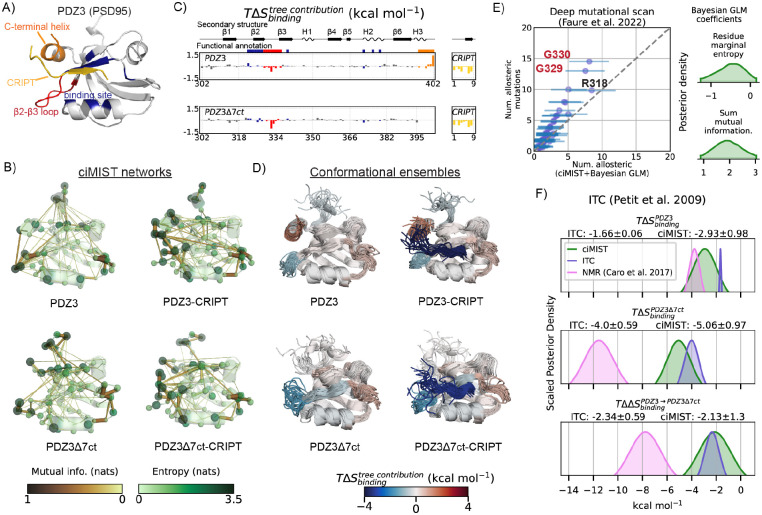
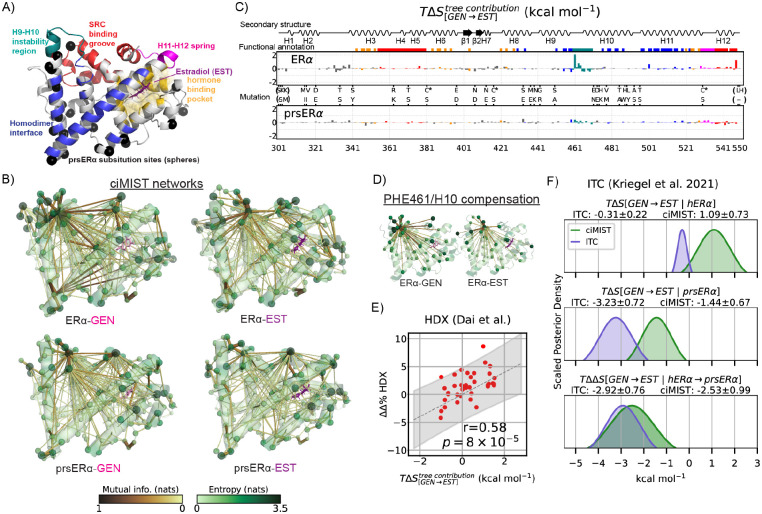
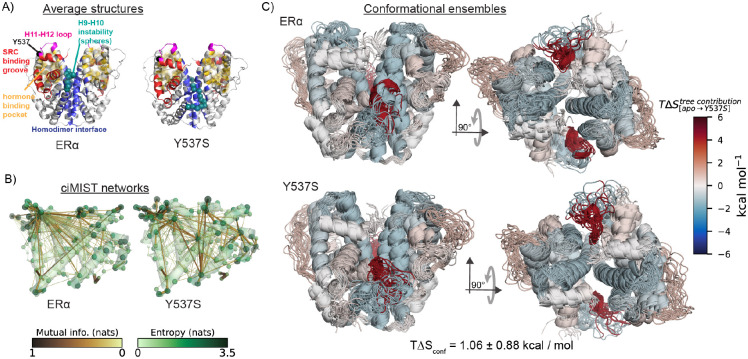
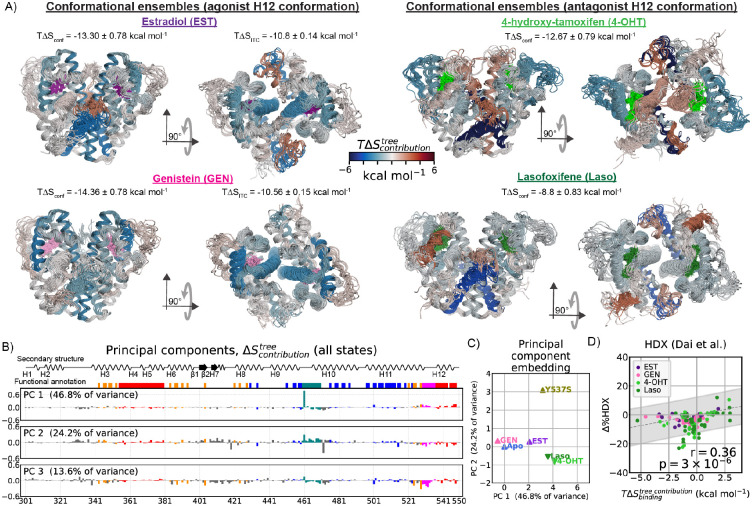
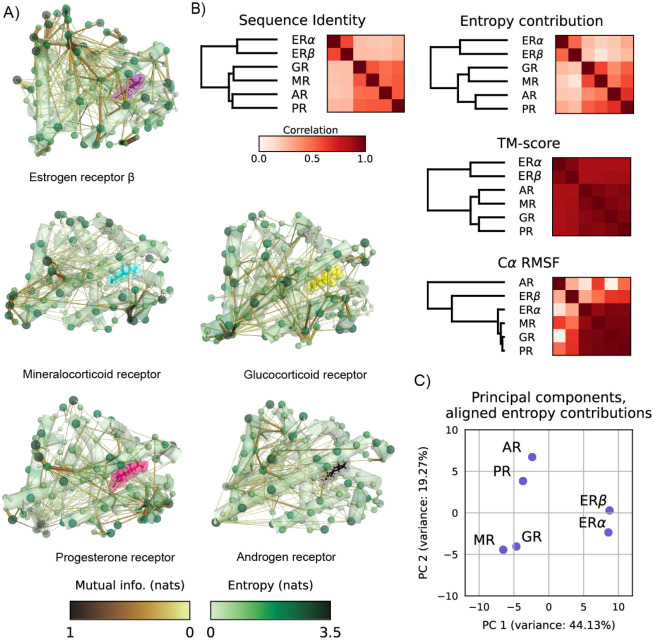
Proteins can sense signals and-in a process called allostery-transmit information to distant sites. Such information is often not encoded by a protein's average structure, but rather by its dynamics in a way that remains unclear. We show that maximum information tree networks learned from microseconds-long molecular dynamics simulations provide mechanistically-detailed maps of information transmission within proteins in a ligand- and mutation-sensitive manner. On a PDZ domain and the entire human steroid receptor family, these networks quantitatively predict functionally relevant experimental datasets spanning multiple scales, including allosteric sensitivity across a saturation mutagenesis library, calorimetric binding entropies, and phylogenetic trees. These results suggest that a sparse network of entropic couplings encodes the dynamics-to-function map; functional reprogramming and diversification by ligand binding and evolution can modify this network without changing protein structure.
Conflict of interest statement
Competing interests: Authors declare that they have no competing interests.
Figures
Similar articles
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
Idiopathic (Genetic) Generalized Epilepsy.2024 Feb 12. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Feb 12. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31536218 Free Books & Documents.
-
Psychological therapies for panic disorder with or without agoraphobia in adults: a network meta-analysis.Cochrane Database Syst Rev. 2016 Apr 13;4(4):CD011004. doi: 10.1002/14651858.CD011004.pub2. Cochrane Database Syst Rev. 2016. PMID: 27071857 Free PMC article.
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
The Lived Experience of Autistic Adults in Employment: A Systematic Search and Synthesis.Autism Adulthood. 2024 Dec 2;6(4):495-509. doi: 10.1089/aut.2022.0114. eCollection 2024 Dec. Autism Adulthood. 2024. PMID: 40018061 Review.
References
-
- Hsieh C., Santell R., Haslam S., Helferich W., Estrogenic effects of genistein on the growth of estrogen receptor-positive human breast cancer (MCF-7) cells in vitro and in vivo. Cancer Research 58, 3833–3838 (1998). - PubMed
-
- Hewitt S., Couse J., Korach K., Estrogen receptor transcription and transactivation Estrogen receptor knockout mice: what their phenotypes reveal about mechanisms of estrogen action. Breast Cancer Research 2, 345 (2000), doi: 10.1186/bcr79, https://doi.org/10.1186/bcr79. - DOI - DOI - PMC - PubMed
-
- Dai S., et al. , Prediction of the tissue-specificity of selective estrogen receptor modulators by using a single biochemical method. Proceedings of the National Academy of Sciences 105, 7171–7176 (2008), publisher: Proceedings of the National Academy of Sciences, doi: 10.1073/pnas.0710802105, https://www.pnas.org/doi/full/10.1073/pnas.0710802105. - DOI - DOI - PMC - PubMed
-
- Fanning S., et al. , Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function- 2 binding conformation. eLife 5, e12792 (2016), publisher: eLife Sciences Publications, Ltd, doi: 10.7554/eLife.12792, https://doi.org/10.7554/eLife.12792. - DOI - DOI - PMC - PubMed
-
- Katzenellenbogen J., Mayne C., Katzenellenbogen B., Greene G., Chandarlapaty S., Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nature Reviews Cancer 18, 377–388 (2018), number: 6 Publisher: Nature Publishing Group, doi: 10.1038/s41568-018-0001-z, https://www.nature.com/articles/s41568-018-0001-z. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources