

This is a preprint.
De novo design of miniprotein agonists and antagonists targeting G protein-coupled receptors
- PMID: 40501737
- PMCID: PMC12157396
- DOI: 10.1101/2025.03.23.644666
De novo design of miniprotein agonists and antagonists targeting G protein-coupled receptors
Abstract
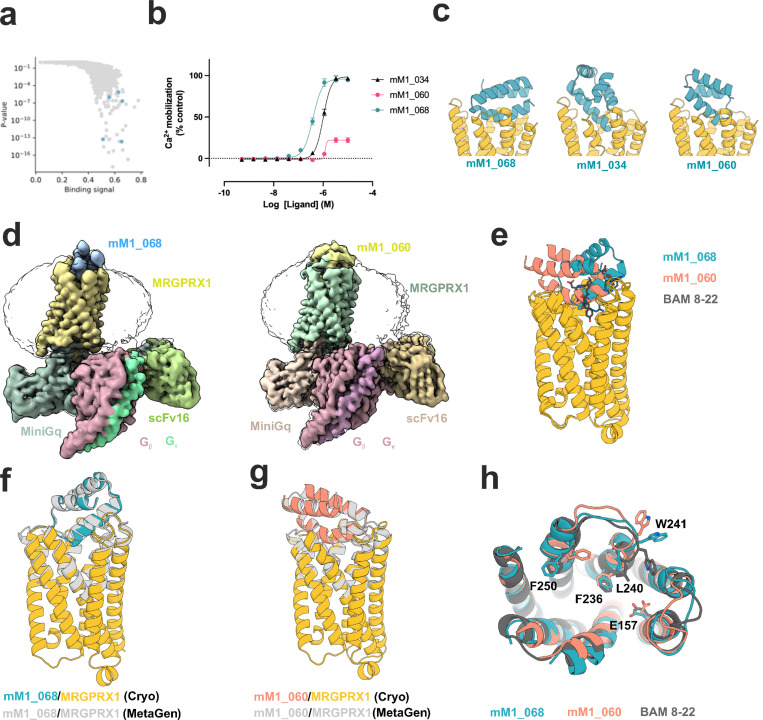
G protein-coupled receptors (GPCRs) play key roles in physiology and are central targets for drug discovery and development, yet the design of protein agonists and antagonists has been challenging as GPCRs are integral membrane proteins and conformationally dynamic. Here we describe computational de novo design methods and a high throughput "receptor diversion" microscopy-based screen for generating GPCR binding miniproteins with high affinity, potency and selectivity, and the use of these methods to generate MRGPRX1 agonists and CXCR4, GLP1R, GIPR, GCGR and CGRPR antagonists. Cryo-electron microscopy data reveals atomic-level agreement between designed and experimentally determined structures for CGRPR-bound antagonists and MRGPRX1-bound agonists, confirming precise conformational control of receptor function. Our de novo design and screening approach opens new frontiers in GPCR drug discovery and development.
Figures




References
-
- Congreve M., De Graaf C., Swain N. A. & Tate C. G. Impact of GPCR Structures on Drug Discovery. Cell 181, 81–91 (2020). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources