

This is a preprint.
Pangenome-aware DeepVariant
- PMID: 40501862
- PMCID: PMC12157594
- DOI: 10.1101/2025.06.05.657102
Pangenome-aware DeepVariant
Abstract
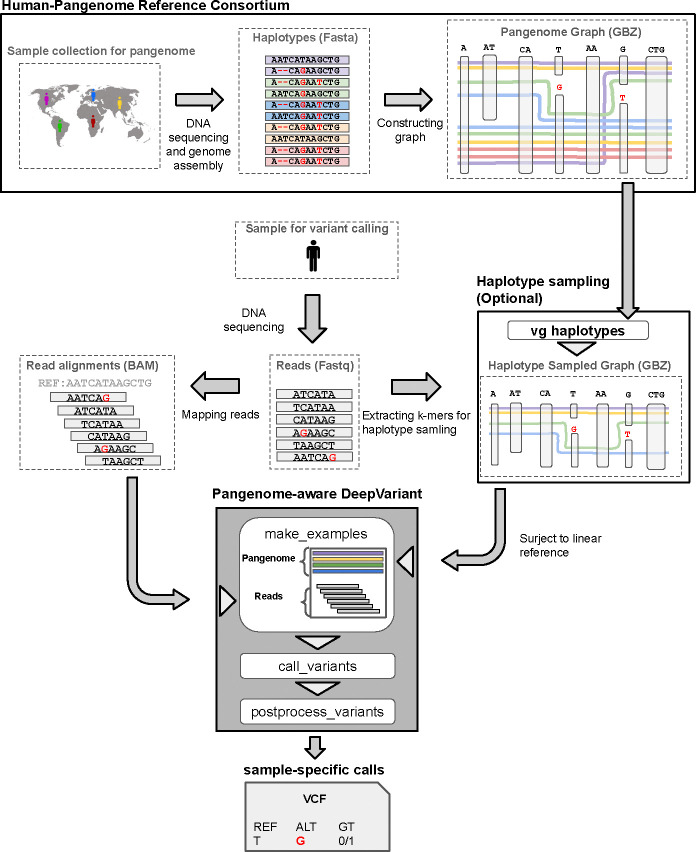
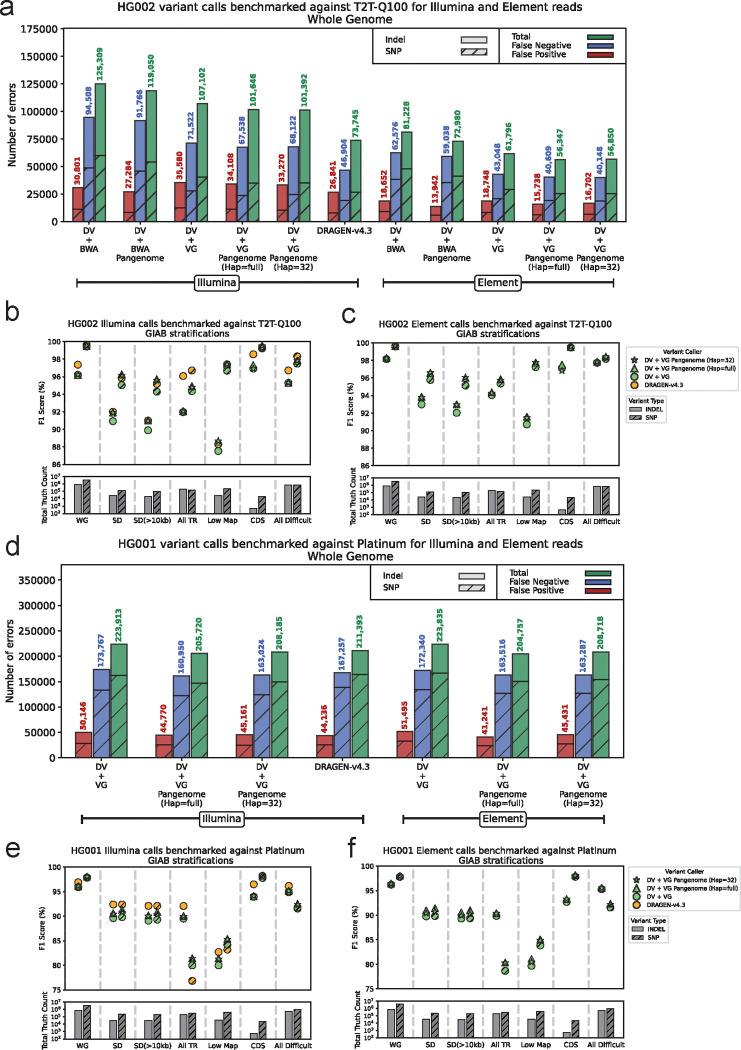
Population-scale genomics information provides valuable prior knowledge for various genomic analyses, especially variant calling. A notable example of such application is the human pangenome reference released by the Human Pangenome Reference Consortium, which has been shown to improve read mapping and structural variant genotyping. In this work, we introduce pangenome-aware DeepVariant, a variant caller that uses a pangenome reference alongside sample-specific read alignments. It generates pileup images of both reads and pangenome haplotypes near potential variants and uses a Convolutional Neural Network to infer genotypes. This approach allows directly using a pangenome for distinguishing true variant signals from sequencing or alignment noise. We assessed its performance on various short-read sequencing platforms and read mappers. Across all settings, pangenome-aware DeepVariant outperformed the linear-reference-based DeepVariant, reducing errors by up to 25.5%. We also show that Element reads with pangenome-aware DeepVariant can achieve 23.6% more accurate variant calling performance compared to existing methods.
Conflict of interest statement
Competing interest statement A.C., D.E.C., P.C., L.D., D.R.W., A.K., J.C.M., L.B., and K.S. are employees of Google LLC and own Alphabet stock as part of the standard compensation package. M.A. was an intern at Google LLC during the study. P.E. was an intern at Roche during the study.
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources