

This is a preprint.
A haplotype-resolved view of human gene regulation
- PMID: 40501892
- PMCID: PMC12157683
- DOI: 10.1101/2024.06.14.599122
A haplotype-resolved view of human gene regulation
Abstract
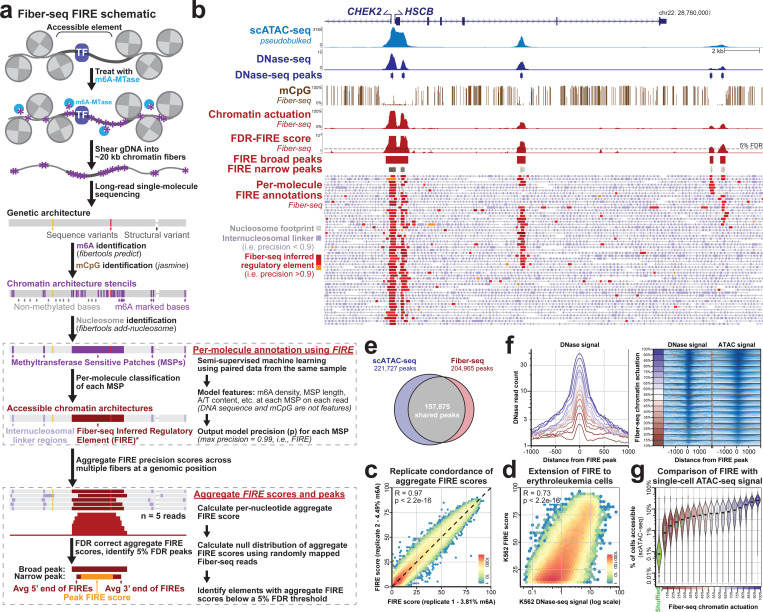
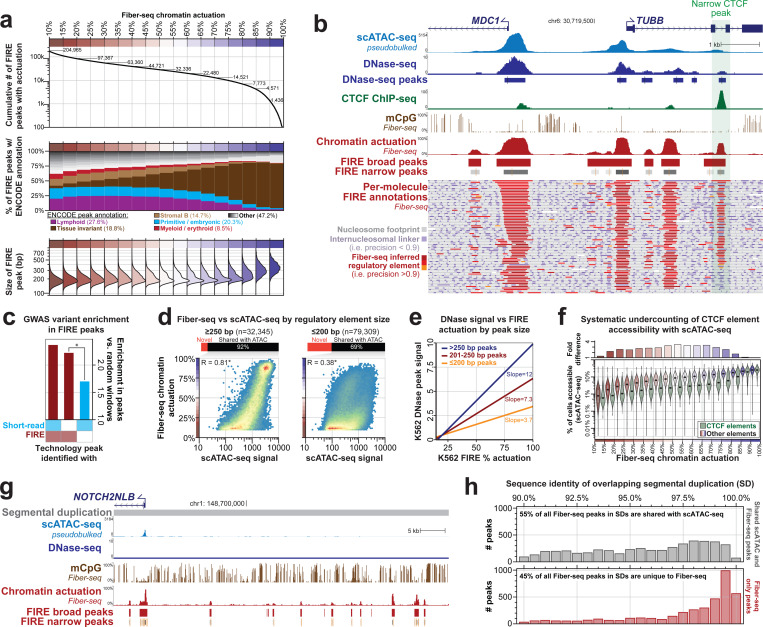
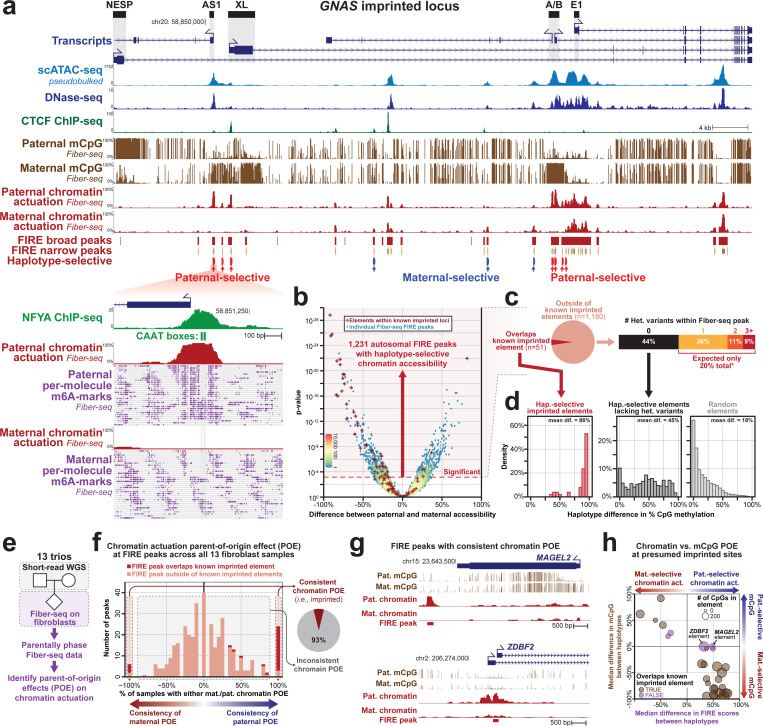
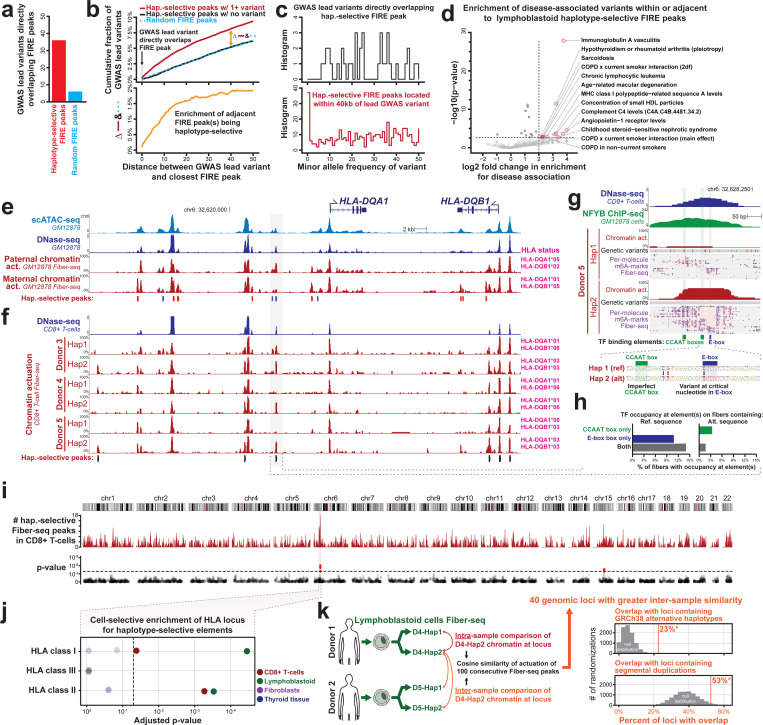
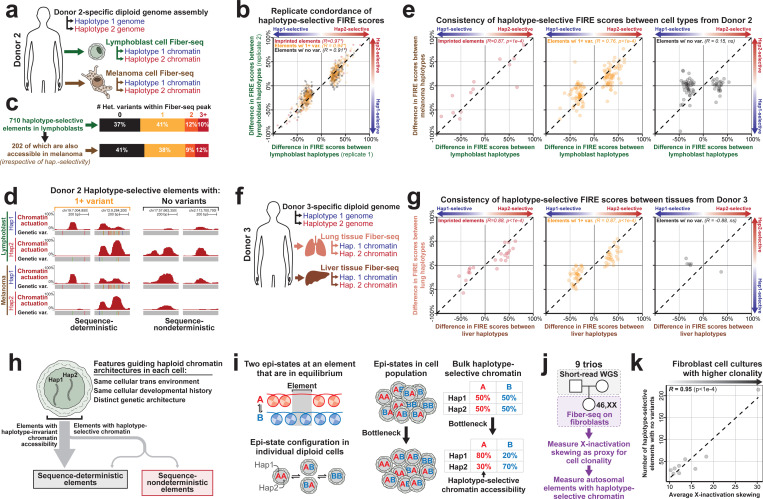
Diploid human cells contain two non-identical genomes, and differences in their regulation underlie human development and disease. We present Fiber-seq Inferred Regulatory Elements (FIRE) and show that FIRE provides a more comprehensive and quantitative snapshot of the accessible chromatin landscape across the 6 Gbp diploid human genome, overcoming previously known and unknown biases afflicting our existing regulatory element catalog. FIRE provides a comprehensive genome-wide map of haplotype-selective chromatin accessibility (HSCA), exposing novel imprinted elements that lack underlying parent-of-origin CpG methylation differences, common and rare genetic variants that disrupt gene regulatory patterns, gene regulatory modules that enable genes to escape X chromosome inactivation, and autosomal mitotically stable somatic epimutations. We find that the human leukocyte antigen (HLA) locus harbors the most HSCA in immune cells, and we resolve the specific transcription factor (TF) binding events disrupted by disease-associated variants within the HLA locus. Finally, we demonstrate that the regulatory landscape of a cell is littered with autosomal somatic epimutations that are propagated by clonal expansions to create mitotically stable and non-genetically deterministic chromatin alterations.
Conflict of interest statement
Competing interests A.B.S. is a co-inventor on a patent relating to the Fiber-seq method (US17/995,058).
Figures

References
-
- Wenger A. M., Peluso P., Rowell W. J., Chang P.-C., Hall R. J., Concepcion G. T., Ebler J., Fungtammasan A., Kolesnikov A., Olson N. D., Töpfer A., Alonge M., Mahmoud M., Qian Y., Chin C.-S., Phillippy A. M., Schatz M. C., Myers G., DePristo M. A., Ruan J., Marschall T., Sedlazeck F. J., Zook J. M., Li H., Koren S., Carroll A., Rank D. R., Hunkapiller M. W., Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol., doi: 10.1038/s41587-019-0217-9 (2019). - DOI - PMC - PubMed
-
- Miga K. H., Koren S., Rhie A., Vollger M. R., Gershman A., Bzikadze A., Brooks S., Howe E., Porubsky D., Logsdon G. A., Schneider V. A., Potapova T., Wood J., Chow W., Armstrong J., Fredrickson J., Pak E., Tigyi K., Kremitzki M., Markovic C., Maduro V., Dutra A., Bouffard G. G., Chang A. M., Hansen N. F., Wilfert A. B., Thibaud-Nissen F., Schmitt A. D., Belton J.-M., Selvaraj S., Dennis M. Y., Soto D. C., Sahasrabudhe R., Kaya G., Quick J., Loman N. J., Holmes N., Loose M., Surti U., Risques R. A., Graves Lindsay T. A., Fulton R., Hall I., Paten B., Howe K., Timp W., Young A., Mullikin J. C., Pevzner P. A., Gerton J. L., Sullivan B. A., Eichler E. E., Phillippy A. M., Telomere-to-telomere assembly of a complete human X chromosome. Nature 585, 79–84 (2020). - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous