

This is a preprint.
Increasing mass spectrometry throughput using time-encoded sample multiplexing
- PMID: 40501910
- PMCID: PMC12154682
- DOI: 10.1101/2025.05.22.655515
Increasing mass spectrometry throughput using time-encoded sample multiplexing
Abstract
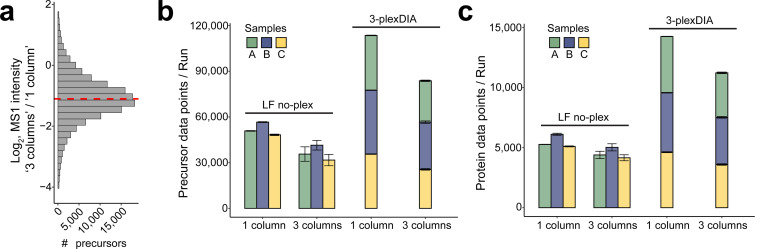
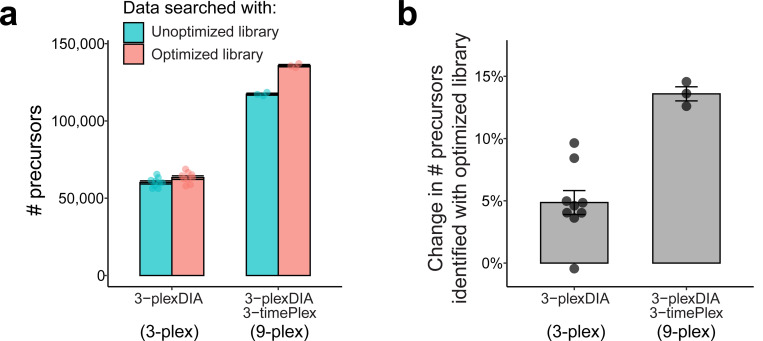
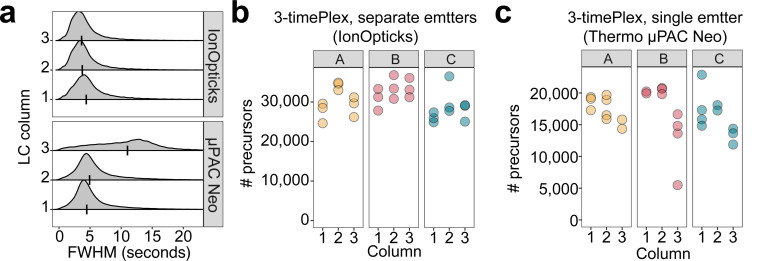
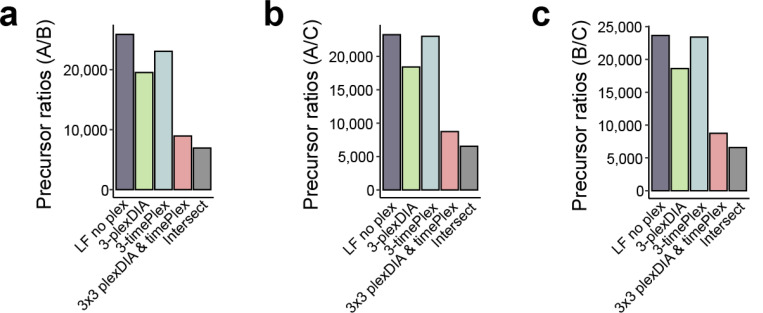
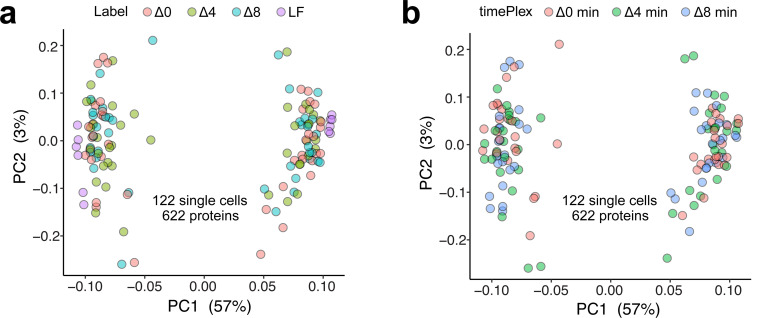
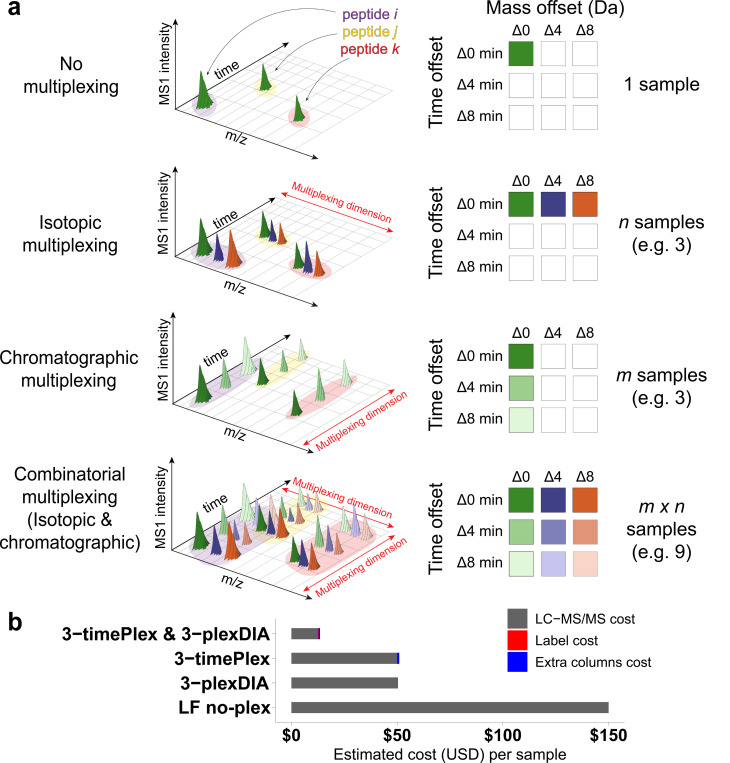
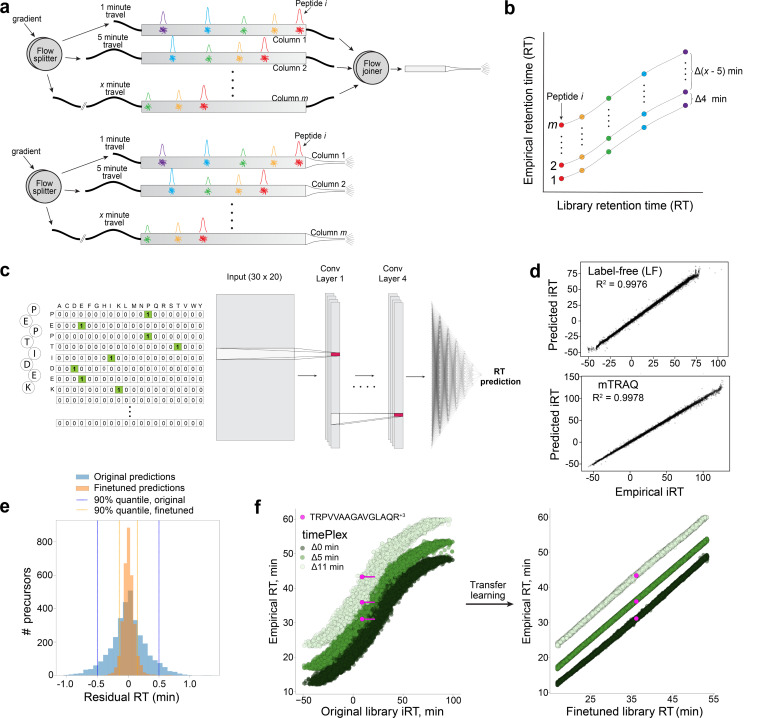
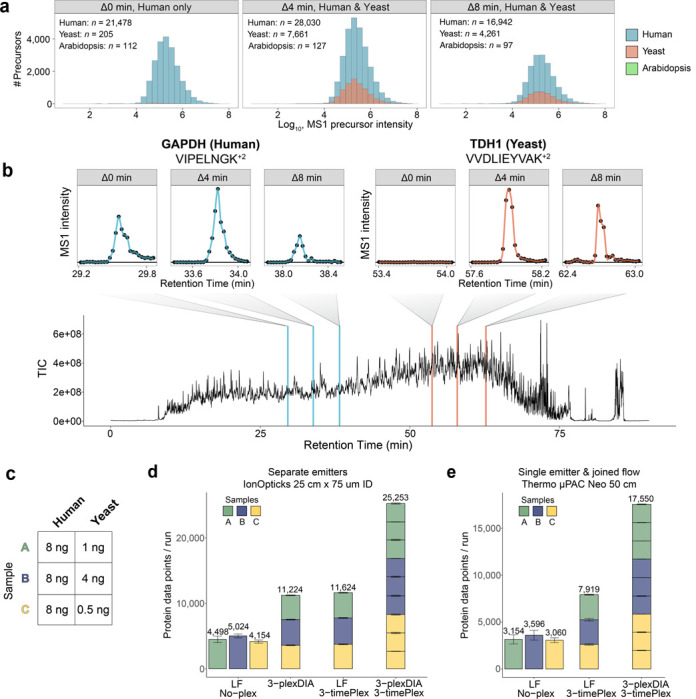
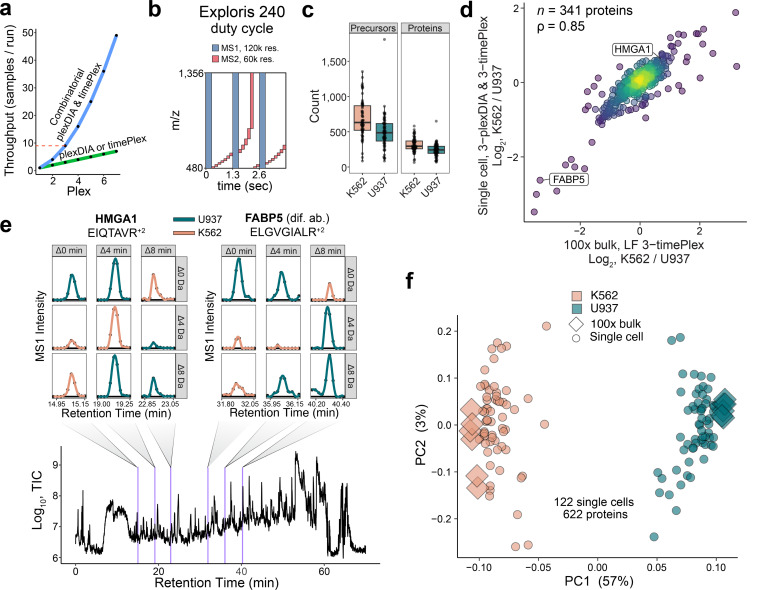
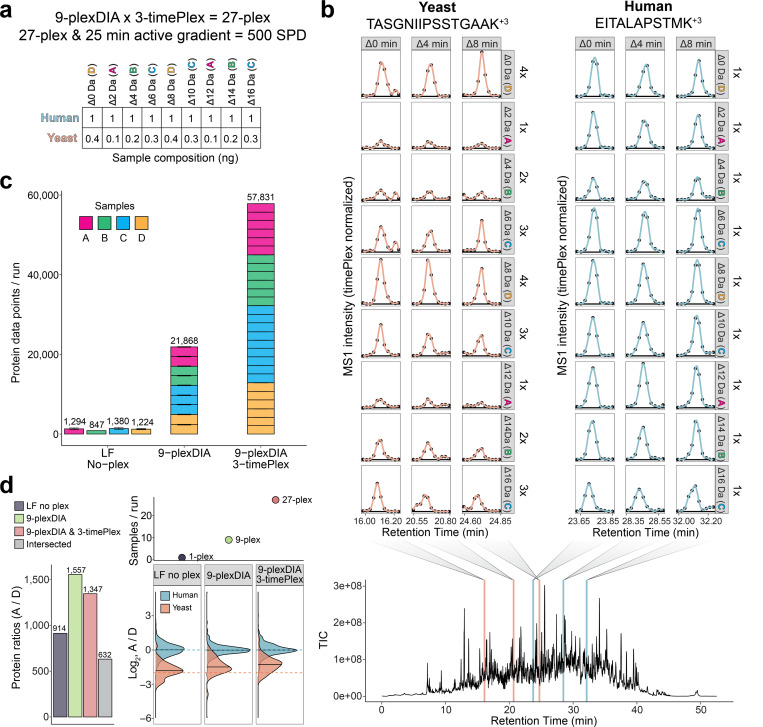
Liquid chromatography-mass spectrometry (LC-MS) can enable precise and accurate quantification of analytes at high-sensitivity, but the rate at which samples can be analyzed remains limiting. Throughput can be increased by multiplexing samples in the mass domain with plexDIA, yet multiplexing along one dimension will only linearly scale throughput with plex. To enable combinatorial-scaling of proteomics throughput, we developed a complementary multiplexing strategy in the time domain, termed 'timePlex'. timePlex staggers and overlaps the separation periods of individual samples. This strategy is orthogonal to isotopic multiplexing, which enables combinatorial multiplexing in mass and time domains when paired together, and thus multiplicatively increased throughput. We demonstrate this with 3-timePlex and 3-plexDIA, enabling the multiplexing of 9 samples per LC-MS run, and 3-timePlex and 9-plexDIA exceeding 500 samples / day with a combinatorial 27-plex. Crucially, timePlex supports sensitive analyses, including of single cells. These results establish timePlex as a methodology for label-free multiplexing and combinatorial scaling of the throughput of LC-MS proteomics. We project this combined approach will eventually enable an increase in throughput exceeding 1,000 samples / day.
Conflict of interest statement
Competing interests: J.D., H.S., K.M.D., and N.S. are listed as inventors on a provisional patent application for timePlex. N.S. is a founding director and CEO of Parallel Squared Technology Institute, which is a nonprofit research institute.
Figures

Similar articles
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
Rapid, point-of-care antigen tests for diagnosis of SARS-CoV-2 infection.Cochrane Database Syst Rev. 2022 Jul 22;7(7):CD013705. doi: 10.1002/14651858.CD013705.pub3. Cochrane Database Syst Rev. 2022. PMID: 35866452 Free PMC article.
-
Interventions targeted at women to encourage the uptake of cervical screening.Cochrane Database Syst Rev. 2021 Sep 6;9(9):CD002834. doi: 10.1002/14651858.CD002834.pub3. Cochrane Database Syst Rev. 2021. PMID: 34694000 Free PMC article.
-
Diagnostic test accuracy and cost-effectiveness of tests for codeletion of chromosomal arms 1p and 19q in people with glioma.Cochrane Database Syst Rev. 2022 Mar 2;3(3):CD013387. doi: 10.1002/14651858.CD013387.pub2. Cochrane Database Syst Rev. 2022. PMID: 35233774 Free PMC article.
-
Behavioral interventions to reduce risk for sexual transmission of HIV among men who have sex with men.Cochrane Database Syst Rev. 2008 Jul 16;(3):CD001230. doi: 10.1002/14651858.CD001230.pub2. Cochrane Database Syst Rev. 2008. PMID: 18646068
References
-
- Nagae N., Itoh H., Nimura N., Kinoshita T. & Takeuchi T. Rapid separation of proteins and peptides by reversed-phase microcolumn liquid chromatography. Journal of Microcolumn Separations 3, 5–9 (1991).
-
- Moritz R. L. & Simpson R. J. Purification of proteins and peptides for sequence analysis using microcolumn liquid chromatography. Journal of Microcolumn Separations 4, 485–489 (1992).
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources