

This is a preprint.
Single-nucleus epigenomic dysregulation unmasks genetic risk-associated neurodegenerative glia states
- PMID: 40502140
- PMCID: PMC12157633
- DOI: 10.1101/2025.06.02.657512
Single-nucleus epigenomic dysregulation unmasks genetic risk-associated neurodegenerative glia states
Abstract
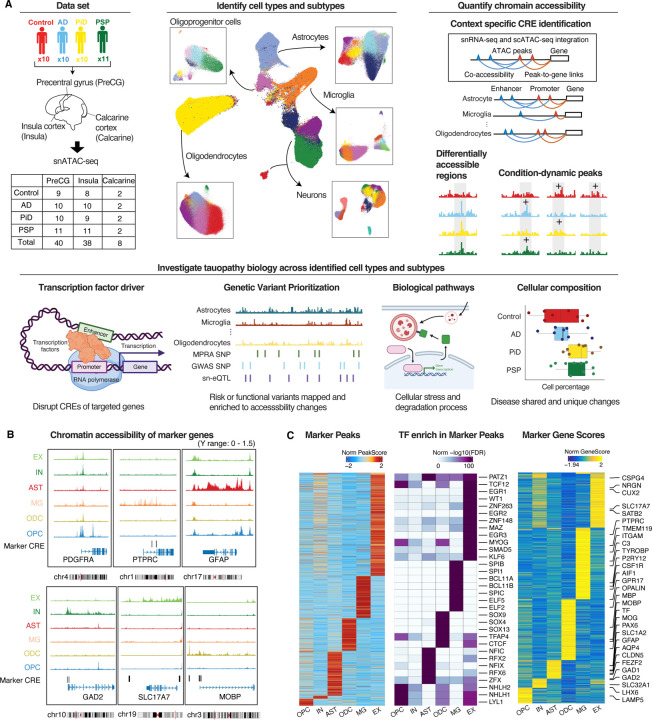
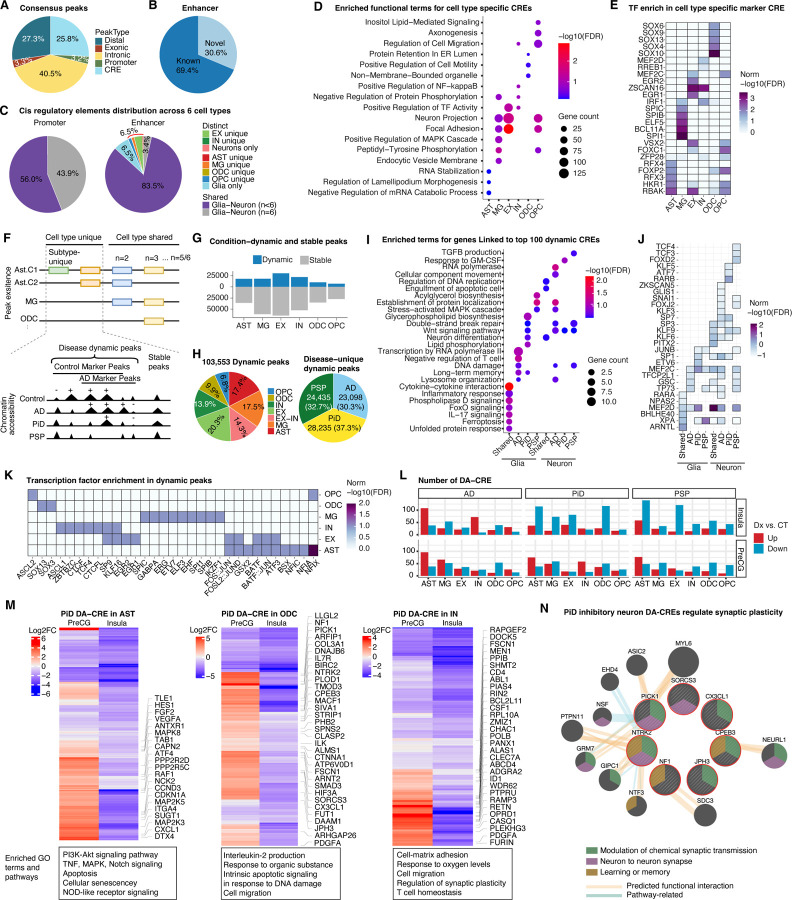
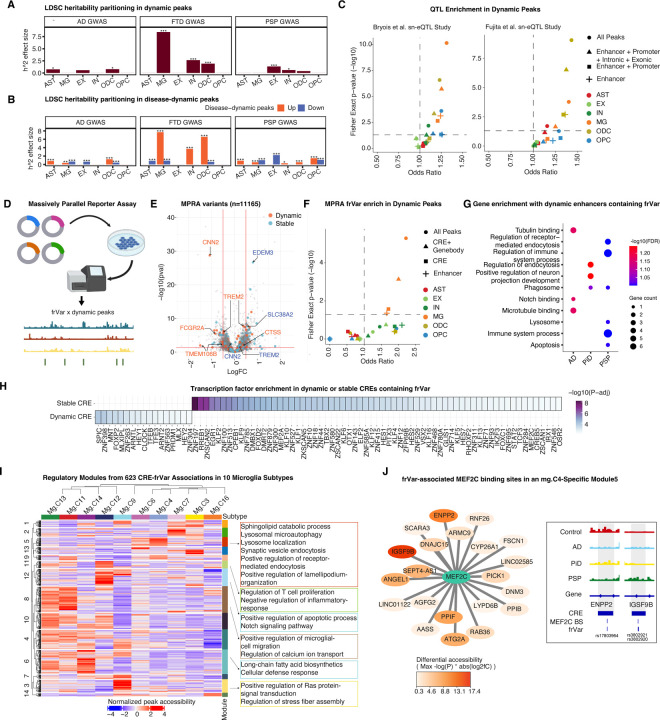
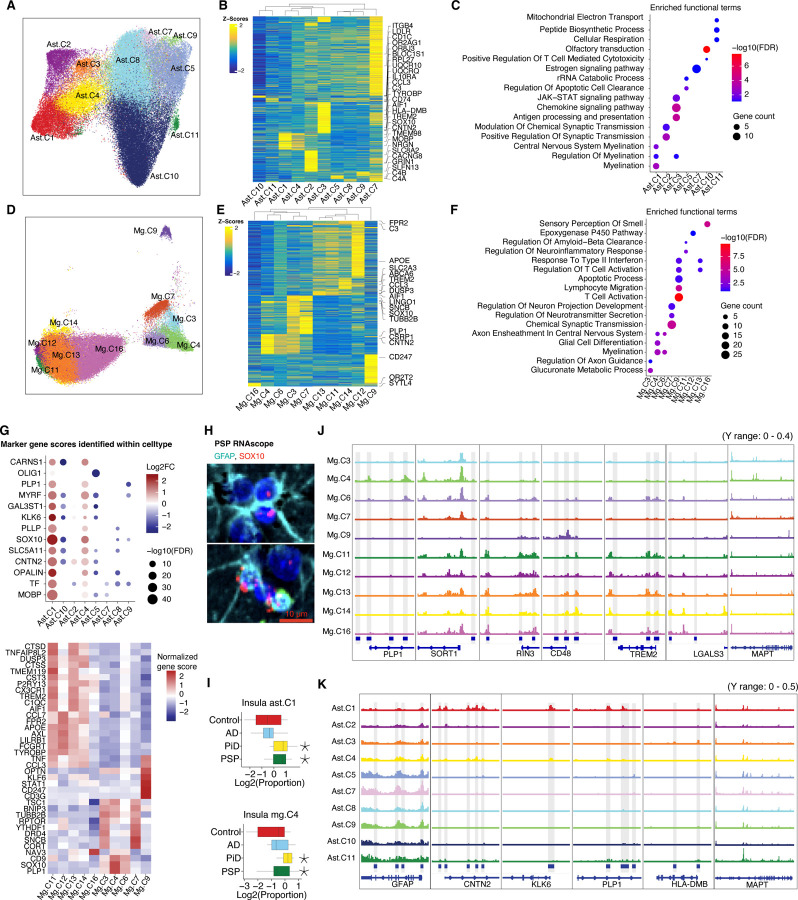
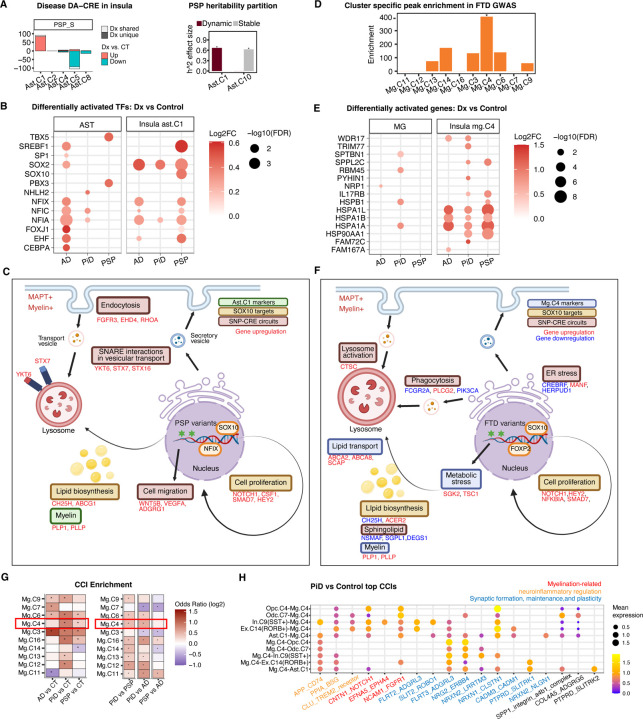
The accumulation of abnormal tau protein selectively affects distinct brain regions and specific populations of neurons and glial cells in tau-related dementias, such as Alzheimer's disease (AD), Pick's disease (PiD), and progressive supranuclear palsy (PSP). Although the three disorders share the feature of tau protein pathology, the regulatory circuitry of non-coding genetic variants underlying risk-associated cell states remains to be elucidated. Using paired single-nucleus profiling of chromatin accessibility and gene expression across AD, PiD, and PSP, we define cell-type-specific cis-regulatory elements (CREs) across six cell types and fifty subclasses. Comparing disease-dynamic CREs across three disorders, we find that glia overrepresent disorder-specific gene regulation related to dynamic cellular response to stress. We show that human genetic variants affecting microglial gene regulation converge into distinct and co-regulated modules affecting specific cellular functions. Moreover, polygenic risk modifiers are maximally co-accessible in disorder-specific glial states, modifying distinct pathways such as sphingomyelin regulation in PiD. Our study informs glial regulators linked to polygenic modifiers of primary tauopathy, introducing modifiable pathways governing resilience for therapeutic consideration.
Conflict of interest statement
Ethics declarations The authors declare no competing interests.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous