

This is a preprint.
An improved model for prediction of de novo designed proteins with diverse geometries
- PMID: 40502157
- PMCID: PMC12157515
- DOI: 10.1101/2025.06.02.657515
An improved model for prediction of de novo designed proteins with diverse geometries
Abstract
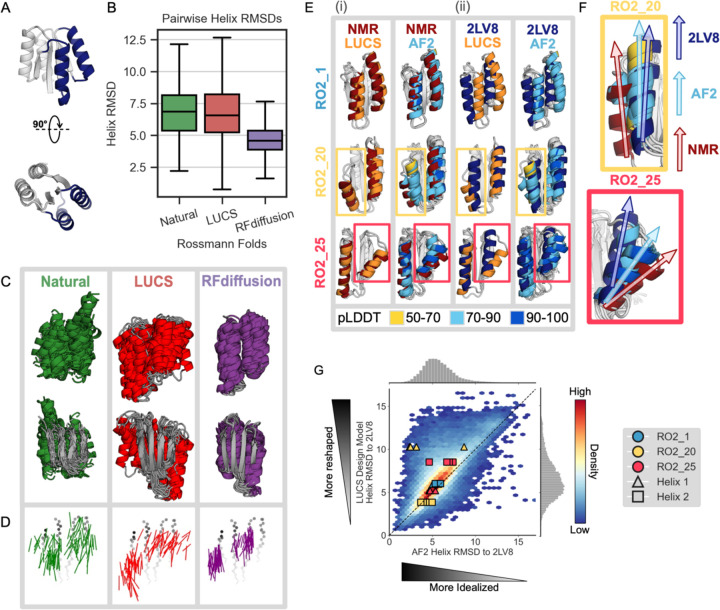
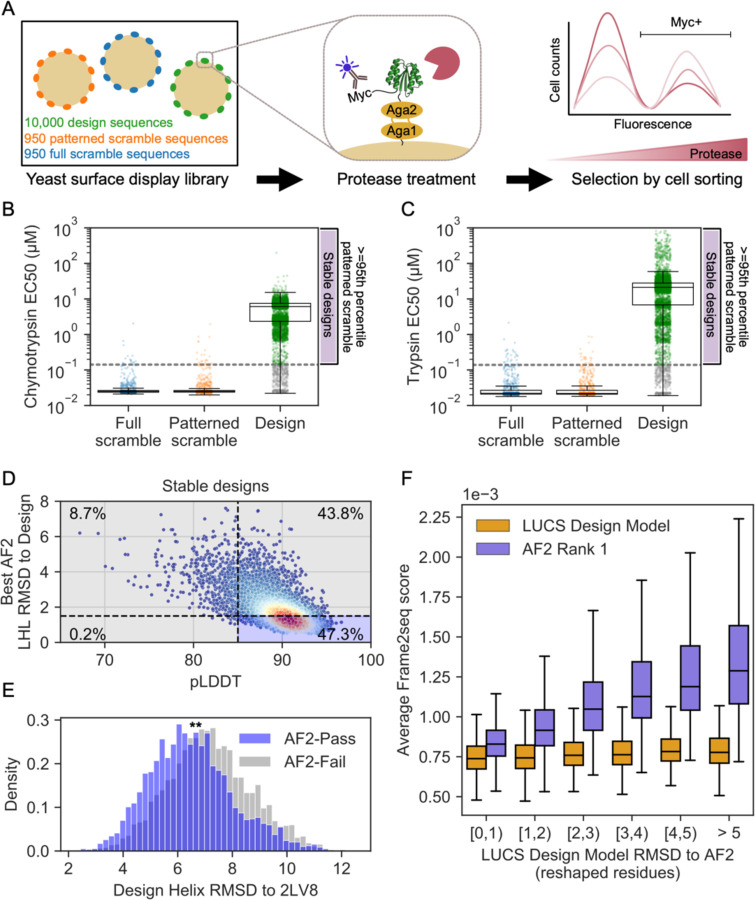
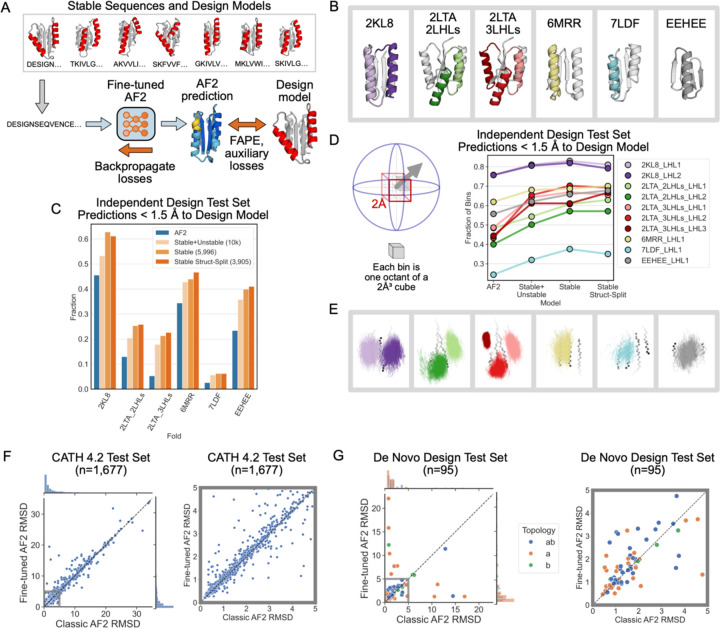
Nature uses structural variations on protein folds to fine-tune the geometries of proteins for diverse functions, yet deep learning-based de novo protein design methods generate highly regular, idealized protein fold geometries that fail to capture natural diversity. Here, using physics-based design methods, we generated and experimentally validated a dataset of 5,996 stable, de novo designed proteins with diverse non-ideal geometries. We show that deep learning-based structure prediction methods applied to this set have a systematic bias towards idealized geometries. To address this problem, we present a fine-tuned version of Alphafold2 that is capable of recapitulating geometric diversity and generalizes to a new dataset of thousands of geometrically diverse de novo proteins from 5 fold families unseen in fine-tuning. Our results suggest that current deep learning-based structure prediction methods do not capture some of the physics that underlie the specific conformational preferences of proteins designed de novo and observed in nature. Ultimately, approaches such as ours and further informative datasets should lead to improved models that reflect more of the physical principles of atomic packing and hydrogen bonding interactions and enable improved generalization to more challenging design problems.
Conflict of interest statement
Competing interests: Authors declare that they have no competing interests.
Figures
References
-
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P & Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 5G6, 583–589 (2021). 10.1038/s41586-021-03819-2 - DOI - PMC - PubMed
-
- Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, Ronneberger O, Willmore L, Ballard AJ, Bambrick J, Bodenstein SW, Evans DA, Hung CC, O’Neill M, Reiman D, Tunyasuvunakool K, Wu Z, Zemgulyte A, Arvaniti E, Beattie C, Bertolli O, Bridgland A, Cherepanov A, Congreve M, Cowen-Rivers AI, Cowie A, Figurnov M, Fuchs FB, Gladman H, Jain R, Khan YA, Low CMR, Perlin K, Potapenko A, Savy P, Singh S, Stecula A, Thillaisundaram A, Tong C, Yakneen S, Zhong ED, Zielinski M, Zidek A, Bapst V, Kohli P, Jaderberg M, Hassabis D & Jumper JM. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024). 10.1038/s41586-024-07487-w - DOI - PMC - PubMed
-
- Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Ǫ, Kinch LN, Schaeffer RD, Millan C, Park H, Adams C, Glassman CR, DeGiovanni A, Pereira JH, Rodrigues AV, van Dijk AA, Ebrecht AC, Opperman DJ, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy MK, Dalwadi U, Yip CK, Burke JE, Garcia KC, Grishin NV, Adams PD, Read RJ & Baker D. Accurate prediction of protein structures and interactions using a three-track neural network. Science (2021). 10.1126/science.abj8754 - DOI - PMC - PubMed
-
- Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, Smetanin N, Verkuil R, Kabeli O, Shmueli Y, Dos Santos Costa A, Fazel-Zarandi M, Sercu T, Candido S & Rives A. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 37G, 1123–1130 (2023). 10.1126/science.ade2574 - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources