

A scalable gut epithelial organoid model reveals the genome-wide colonization landscape of a human-adapted pathogen
- PMID: 40506541
- PMCID: PMC12283395
- DOI: 10.1038/s41588-025-02218-x
A scalable gut epithelial organoid model reveals the genome-wide colonization landscape of a human-adapted pathogen
Abstract
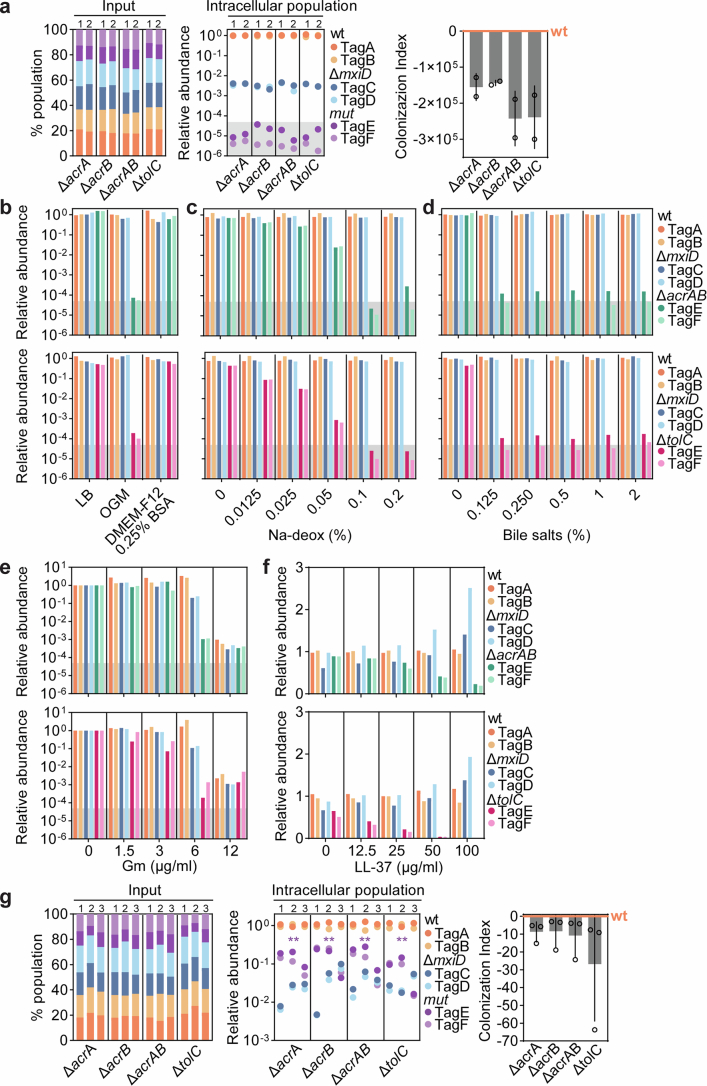
Studying the pathogenesis of human-adapted microorganisms is challenging, since small animal models often fail to recapitulate human physiology. Hence, the comprehensive genetic and regulatory circuits driving the infection process of principal human pathogens such as Shigella flexneri remain to be defined. We combined large-scale Shigella infections of enteroids and colonoids with transposon-directed insertion sequencing and Bayesian statistical modeling to address infection bottlenecks, thereby establishing the comprehensive genome-wide map of Shigella genes required to infect human intestinal epithelium. This revealed the Shigella virulence effectors essential for epithelial cell colonization across geometries and intestinal segments, identified over 100 chromosomal genes involved in the process and uncovered a post-transcriptional mechanism whereby tRNA-modification enzymes and differential codon usage exert global control of a bacterial virulence program. Our findings provide a broadly applicable framework for combining advanced organotypic tissue culture with functional genomics and computational tools to map human-microorganism interactions at scale.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures










References
MeSH terms
Grants and funding
- 2018-02223; 2022-01590/Vetenskapsrådet (Swedish Research Council)
- FFL18-0165/Stiftelsen för Strategisk Forskning (Swedish Foundation for Strategic Research)
- Research Grant 2020/European Society of Clinical Microbiology and Infectious Diseases (ESCMID)
- M21112/Stiftelsen Clas Groschinskys Minnesfond (Clas Groschinski's Memorial Foundation)
- CTS 22:1915/Carl Tryggers Stiftelse för Vetenskaplig Forskning (Carl Trygger Foundation)
LinkOut - more resources
Full Text Sources
