

Aspartate in the Brain: A Review
- PMID: 40506607
- PMCID: PMC12162812
- DOI: 10.1007/s11064-025-04454-3
Aspartate in the Brain: A Review
Abstract
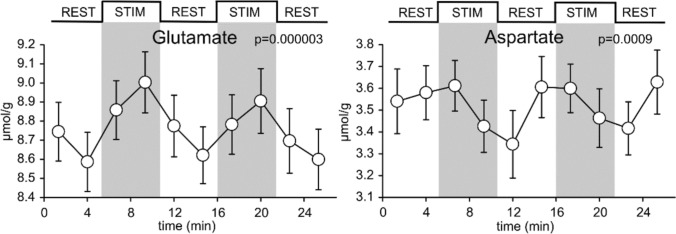
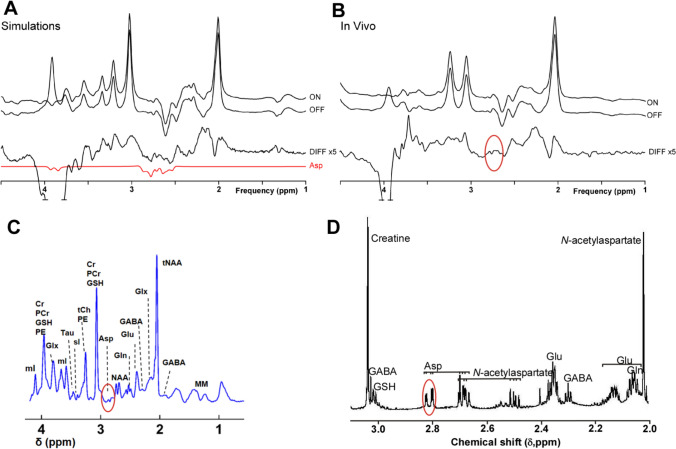
L-Aspartate (aspartic acid; C4H7NO4; 2-aminobutanedoic acid) is a non-essential α-amino acid found ubiquitously throughout the body, including in the brain. Aspartate is one of the protein-forming amino acids and the formation of tRNA-aspartate complex is catalysed by aspartyl tRNA synthetase. Free aspartate, which is the main subject of this review, plays key roles in metabolism, as an amino donor and acceptor. It contributes to the synthesis of protein, arginine and nitric oxide, asparagine, N-acetylaspartate and N-methyl-D-aspartate. Its major metabolic role in the brain is recycling reducing equivalents (protons) between the cytoplasm and mitochondrial matrix as part of the malate-aspartate shuttle. L-Aspartate's actions on synaptic receptors, as well as its possible presence in nerve terminals and synaptic vesicles, are, in principle, consistent with a role as an excitatory neurotransmitter. The evidence is far from conclusive and at times controversial. The role of D-aspartate in brain function is even less certain but, it appears that, rather than being a minor neurotransmitter, D-aspartate is more likely to be involved in fine regulation of endocrine and homeostatic processes. Much research remains to be done in this area. The diversity of its functions and chemistry make aspartate a complex molecule to investigate and measure in vivo. Perturbations of aspartate metabolism have been described in a range of neurological deficits, particularly those of white matter. Here, we examine what is known about the various roles of aspartate in brain, its metabolism, transport and compartmentation, its role as a neurotransmitter or a more general signalling molecule, and what is currently known about its role(s) in disease processes.
Keywords: d-aspartate; Energy metabolism; Malate aspartate shuttle; Neurotransmitter.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing Interests: The authors declare no competing interests.
Figures





References
-
- Liebecq C (1992) Biochemical nomenclature and related documents. Portland Press, London
-
- Vickery HB, Schmidt CL (1931) The history of the discovery of the amino acids. Chem Rev 9(2):169–318
-
- Plisson A (1827) Sur l’identité du malate acide d’althéine avec l’asparagine. Ann chim phys 36:175
-
- Liebig J (1833) Ueber die zusammensetzung des asparamids und der asparaginsäure. Annalen der Pharmacie 7(2):146–150
-
- Kreusler W (1869) Asparaginsäure als Zersetzungsproduct thierischer Proteïnstoffe etc. J Prakt Chem 107(1):240–245
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
