

Elexacaftor/Tezacaftor/Ivacaftor Supports Treatment for CF with ΔI1023-V1024-CFTR
- PMID: 40508114
- PMCID: PMC12155120
- DOI: 10.3390/ijms26115306
Elexacaftor/Tezacaftor/Ivacaftor Supports Treatment for CF with ΔI1023-V1024-CFTR
Abstract
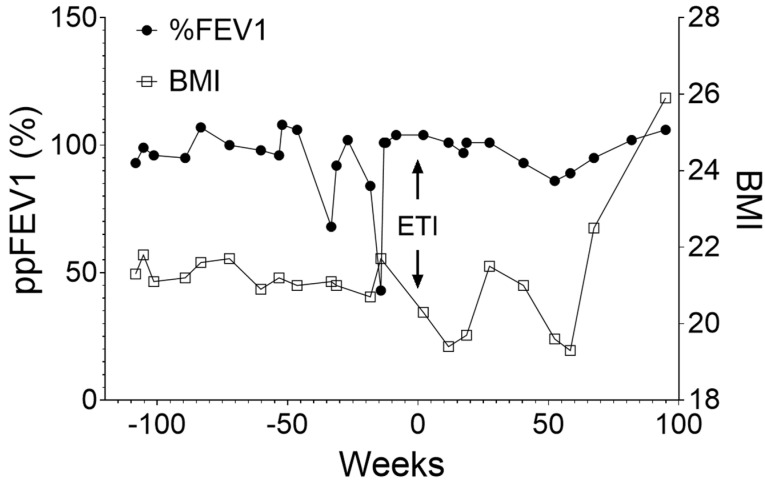
Cystic Fibrosis (CF) is a common genetic disease in the United States, resulting from mutations in the Cystic Fibrosis transmembrane conductance regulator (cftr) gene. CFTR modulators, particularly Elexacaftor/Tezacaftor/Ivacaftor (ETI), have significantly improved clinical outcomes for patients with CF. However, many CFTR mutations are not eligible for CFTR modulator therapy due to their rarity. In this study, we report that a patient carrying rare complex CFTR mutations, c.1680-877G>T and c.3067_3072delATAGTG, showed positive clinical outcomes after ETI treatment. We demonstrate that ETI was able to increase the expression of CFTR harboring c.3067_3072delATAGTG in a heterologous system. Importantly, patient-derived nasal epithelial cells in an air-liquid interface (ALI) culture showed improved CFTR function following ETI treatment. These findings supported the initiation of ETI with the patient. Retrospective studies have suggested that the patient has shown small but steady improvement over the past two years in several clinical metrics, including lung function, body mass index (BMI), and sweat chloride levels. Our studies suggest that ETI could be beneficial for patients carrying c.3067_3072delATAGTG.
Keywords: CFTR; CFTR modulators; Cystic Fibrosis (CF); I1023-V1024; nasal epithelial cells; tTheratyping.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

Similar articles
-
Evaluation of elexacaftor-tezacaftor-ivacaftor treatment in individuals with cystic fibrosis and CFTRN1303K in the USA: a prospective, multicentre, open-label, single-arm trial.Lancet Respir Med. 2024 Dec;12(12):947-957. doi: 10.1016/S2213-2600(24)00205-4. Epub 2024 Aug 26. Lancet Respir Med. 2024. PMID: 39208836
-
Vanzacaftor-tezacaftor-deutivacaftor versus elexacaftor-tezacaftor-ivacaftor in individuals with cystic fibrosis aged 12 years and older (SKYLINE Trials VX20-121-102 and VX20-121-103): results from two randomised, active-controlled, phase 3 trials.Lancet Respir Med. 2025 Mar;13(3):256-271. doi: 10.1016/S2213-2600(24)00411-9. Epub 2025 Jan 2. Lancet Respir Med. 2025. PMID: 39756424 Free PMC article. Clinical Trial.
-
Persistence and evolution of Pseudomonas aeruginosa following initiation of highly effective modulator therapy in cystic fibrosis.mBio. 2024 May 8;15(5):e0051924. doi: 10.1128/mbio.00519-24. Epub 2024 Apr 2. mBio. 2024. PMID: 38564694 Free PMC article.
-
Corrector therapies (with or without potentiators) for people with cystic fibrosis with class II CFTR gene variants (most commonly F508del).Cochrane Database Syst Rev. 2023 Nov 20;11(11):CD010966. doi: 10.1002/14651858.CD010966.pub4. Cochrane Database Syst Rev. 2023. PMID: 37983082 Free PMC article.
-
The impact of cystic fibrosis transmembrane conductance regulator (CFTR) modulators on the pulmonary microbiota.Microbiology (Reading). 2025 Apr;171(4):001553. doi: 10.1099/mic.0.001553. Microbiology (Reading). 2025. PMID: 40202901 Free PMC article. Review.
References
-
- Clancy J.P., Cotton C.U., Donaldson S.H., Solomon G.M., VanDevanter D.R., Boyle M.P., Gentzsch M., Nick J.A., Illek B., Wallenburg J.C., et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019;18:22–34. doi: 10.1016/j.jcf.2018.05.004. - DOI - PMC - PubMed
-
- Dreano E., Burgel P.R., Hatton A., Bouazza N., Chevalier B., Macey J., Leroy S., Durieu I., Weiss L., Grenet D., et al. Theratyping cystic fibrosis patients to guide elexacaftor/tezacaftor/ivacaftor out-of-label prescription. Eur. Respir. J. 2023;62:2300110. doi: 10.1183/13993003.00110-2023. - DOI - PubMed
-
- Kleinfelder K., Lotti V., Eramo A., Amato F., Lo Cicero S., Castelli G., Spadaro F., Farinazzo A., Dell’Orco D., Preato S., et al. In silico analysis and theratyping of an ultra-rare CFTR genotype (W57G/A234D) in primary human rectal and nasal epithelial cells. iScience. 2023;26:108180. doi: 10.1016/j.isci.2023.108180. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
