

Identification of significant genome-wide associations and QTL underlying variation in seed protein composition in pea (Pisum sativum L.)
- PMID: 40511543
- PMCID: PMC12163866
- DOI: 10.1002/tpg2.70051
Identification of significant genome-wide associations and QTL underlying variation in seed protein composition in pea (Pisum sativum L.)
Abstract
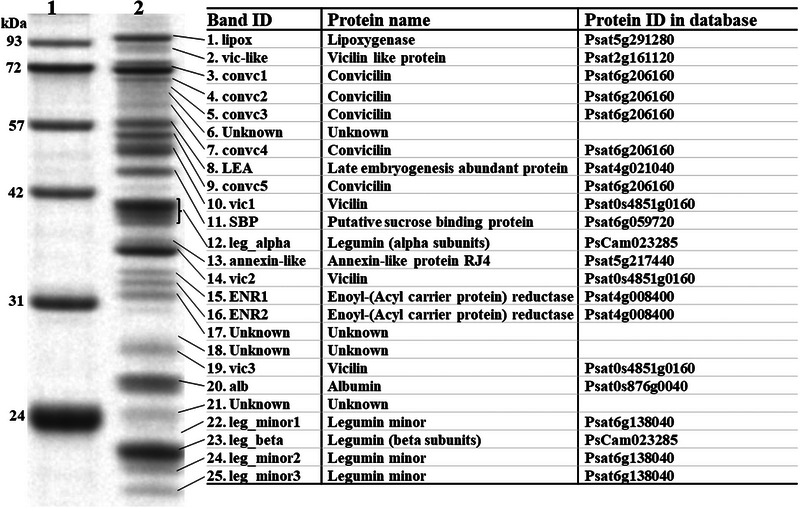
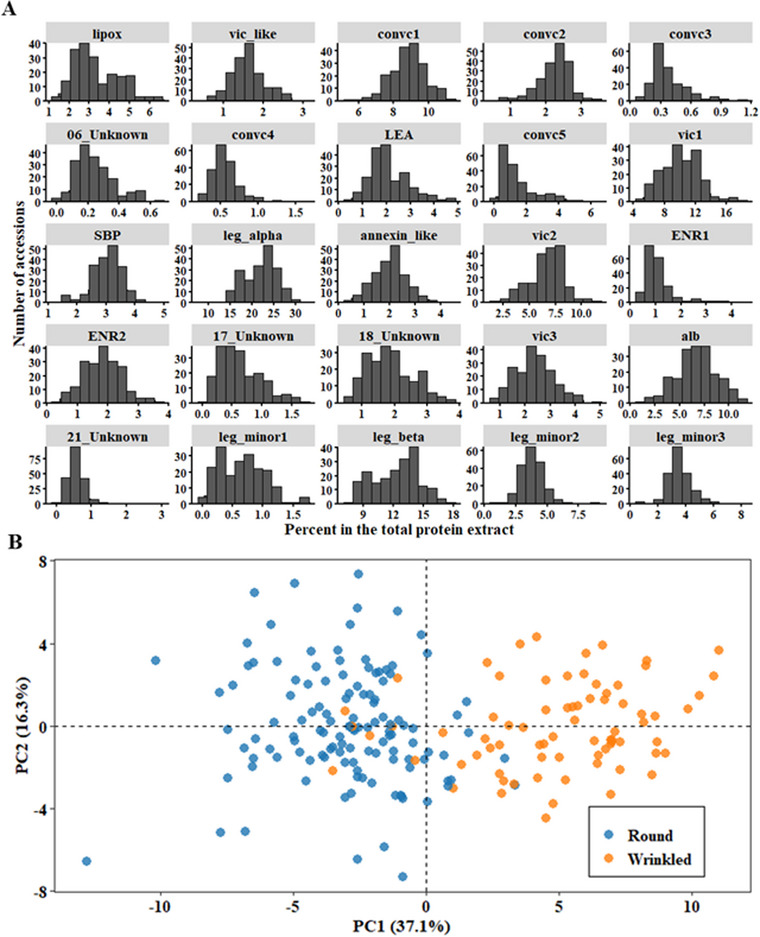
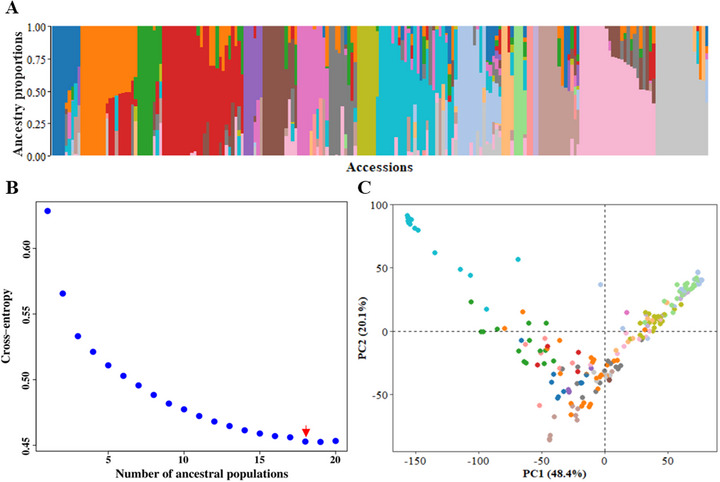
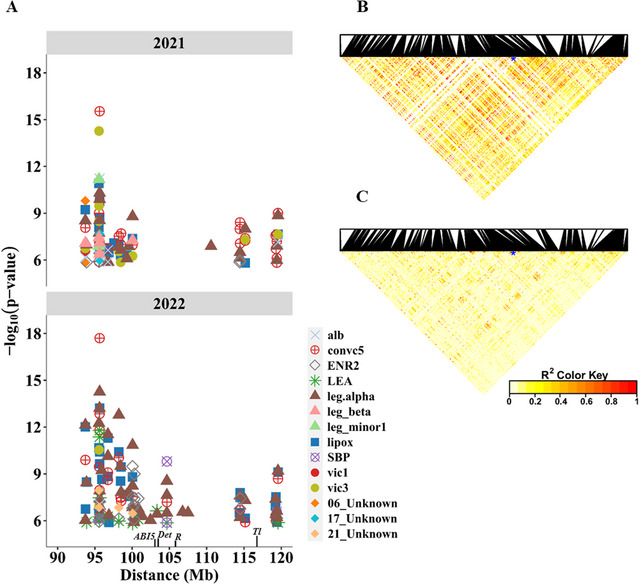
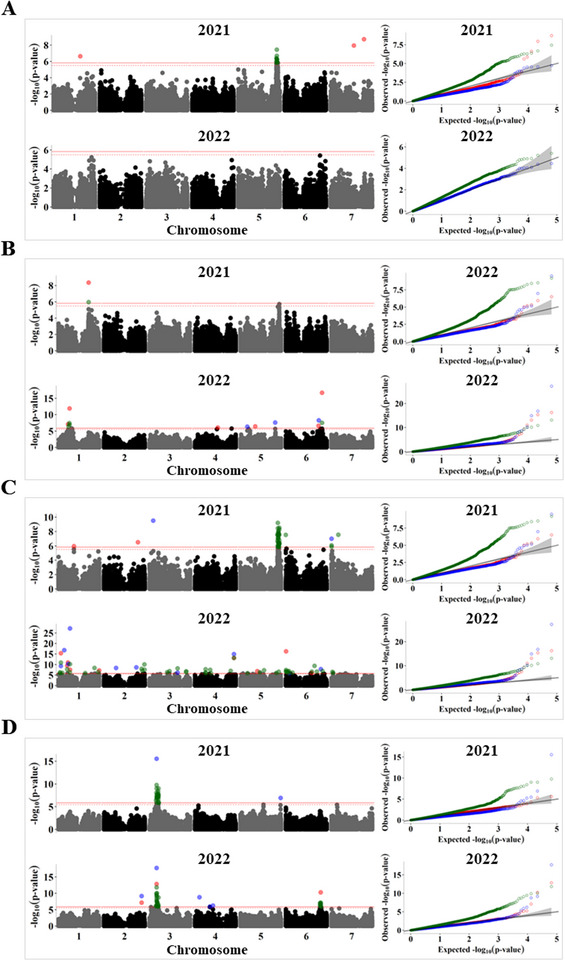
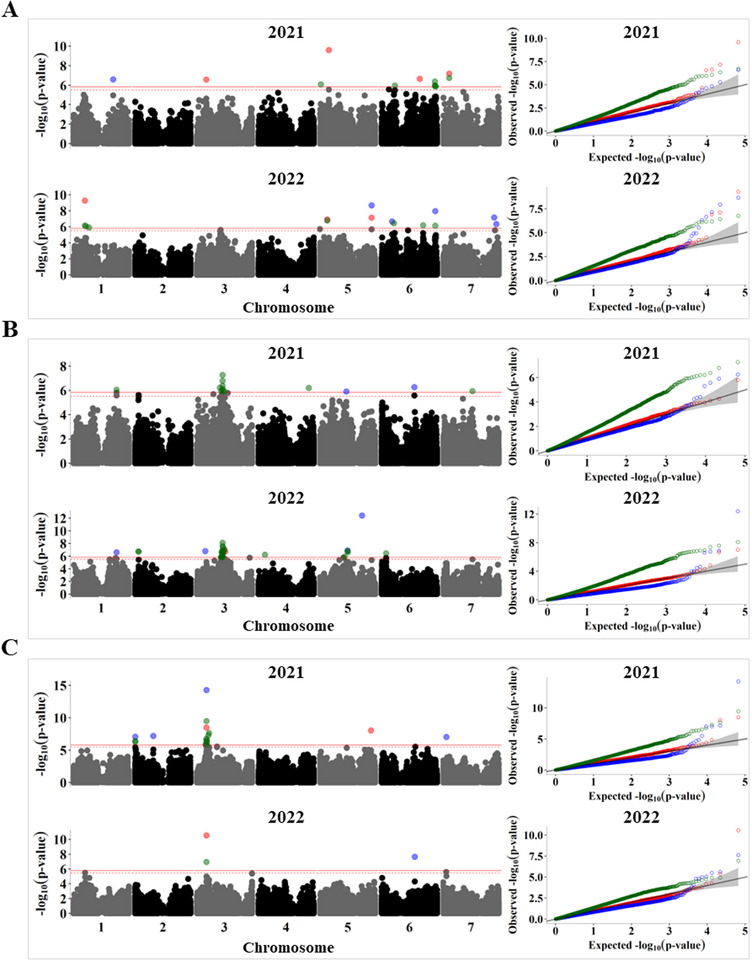
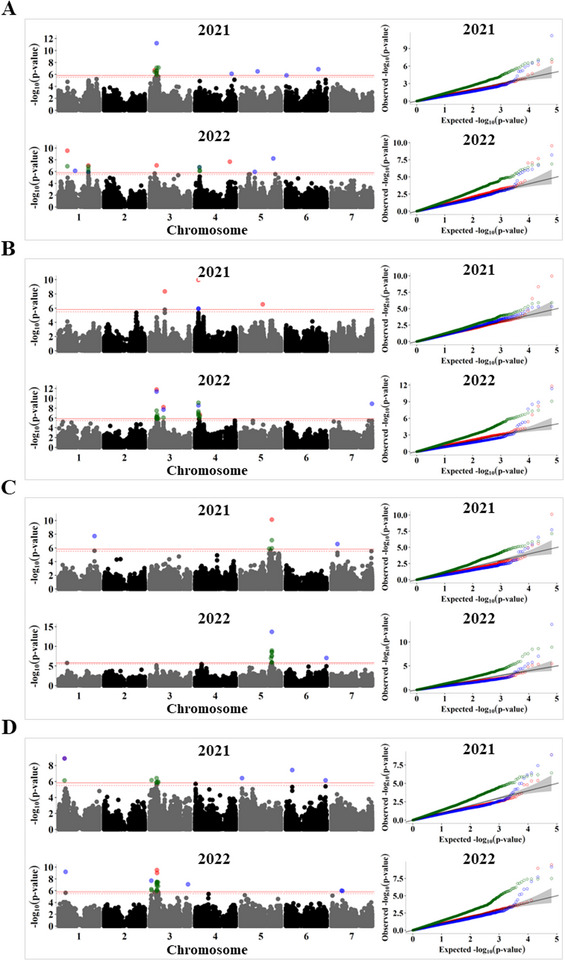
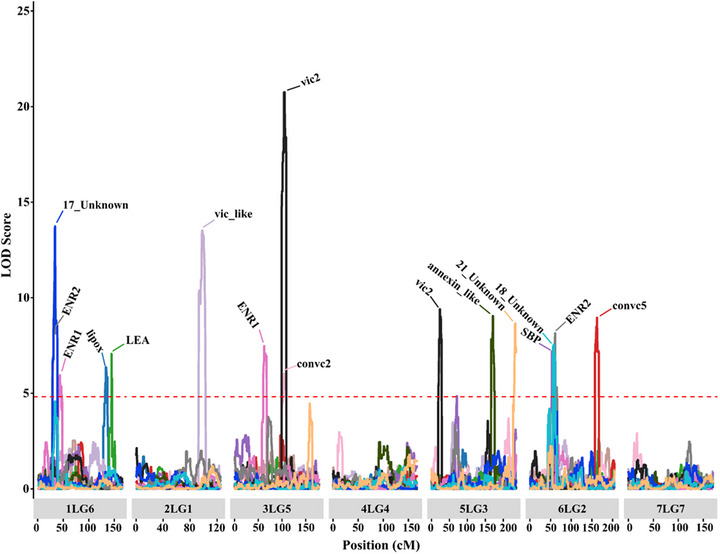
Pulses are a valuable source of plant proteins for human and animal nutrition and have various industrial applications. Understanding the genetic basis for the relative abundance of different seed storage proteins is crucial for developing cultivars with improved protein quality and functional properties. In this study, we employed two complementary approaches, genome-wide association study (GWAS) and quantitative trait locus (QTL) mapping, to identify genetic loci underlying seed protein composition in pea (Pisum sativum L.). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was used to separate the seed proteins, and their relative abundance was quantified using densitometric analysis. For GWAS, we analyzed a diverse panel of 209 accessions genotyped with an 84,691 single-nucleotide polymorphism (SNP) array and identified genetic loci significantly associated with globulins, such as convicilin, vicilin, legumins, and non-globulins, including lipoxygenase, late embryogenesis abundant protein, and annexin-like protein. Additionally, using QTL mapping with 96 recombinant inbred lines, we mapped 11 QTL, including five that overlapped with regions identified by GWAS for the same proteins. Some of the significant SNPs were within or near the genes encoding seed proteins and other genes with predicted functions in protein biosynthesis, trafficking, and modification. This comprehensive genetic mapping study serves as a foundation for future breeding efforts to improve protein quality in pea and other legumes.
© 2025 The Author(s). The Plant Genome published by Wiley Periodicals LLC on behalf of Crop Science Society of America.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Alves‐Carvalho, S. , Aubert, G. , Carrère, S. , Cruaud, C. , Brochot, A.‐L. , Jacquin, F. , Klein, A. , Martin, C. , Boucherot, K. , Kreplak, J. , da Silva, C. , Moreau, S. , Gamas, P. , Wincker, P. , Gouzy, J. , & Burstin, J. (2015). Full‐length de novo assembly of RNA‐seq data in pea (Pisum sativum L.) provides a gene expression atlas and gives insights into root nodulation in this species. The Plant Journal, 84(1), 1–19. 10.1111/tpj.12967 - DOI - PubMed
-
- Bhowmik, P. , Yan, W. , Hodgins, C. , Polley, B. , Warkentin, T. , Nickerson, M. , Ro, D. K. , Marsolais, F. , Domoney, C. , Shariati‐Ievari, S. , & Aliani, M. (2023). CRISPR/Cas9‐mediated lipoxygenase gene‐editing in yellow pea leads to major changes in fatty acid and flavor profiles. Frontiers in Plant Science, 14, 1246905. 10.3389/fpls.2023.1246905 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
