

The cell wall hydrolase MltG is essential to maintain cell wall homeostasis of Enterococcus faecalis
- PMID: 40511937
- PMCID: PMC12288455
- DOI: 10.1128/jb.00056-25
The cell wall hydrolase MltG is essential to maintain cell wall homeostasis of Enterococcus faecalis
Abstract
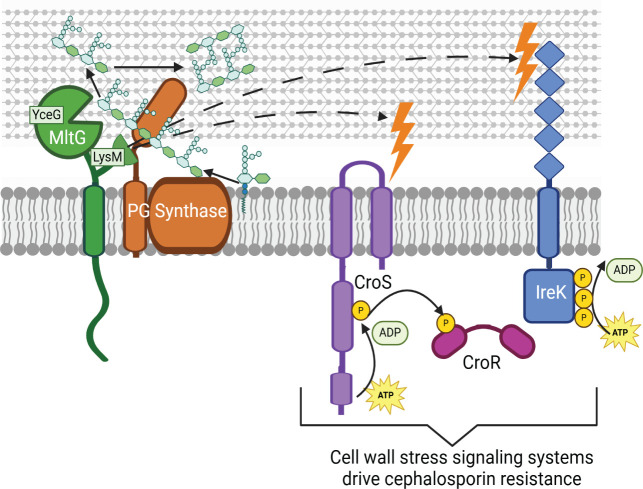
Infections caused by enterococci are increasingly prevalent and difficult to treat due to multidrug resistance. Enterococcus faecalis exhibits intrinsic resistance toward cephalosporins, which inhibit the final step of peptidoglycan (PG) synthesis. Intrinsic resistance requires multiple factors in the PG synthesis pathway and at least two cell-wall-stress signal transduction systems; however, the complete molecular mechanism of enterococcal cephalosporin resistance remains to be elucidated. MltG, a predicted PG hydrolase, is thought to process nascent strands of PG, suggesting that MltG might play an important role in enterococcal cell wall homeostasis and potentially cephalosporin resistance. Here, we demonstrate that enterococcal MltG cleaves nascent PG. An E. faecalis mutant lacking MltG exhibits several related phenotypes in the absence of exogenous stress: a marked growth defect, a loss of cell wall integrity, a reduction in PG synthesis, and activation of two cell-wall-stress signal transduction systems that drive elevated cephalosporin resistance. Together, these results are consistent with the model that MltG promotes proper cell wall homeostasis in E. faecalis, and further reveal that the enzymatic activity of MltG is not necessary for it to perform this function-instead, the LysM (putative PG-binding) domain of MltG plays the critical role. Nevertheless, the enzymatic activity of MltG does impact cephalosporin resistance, because a catalytically inactive MltG variant leads to elevated resistance. Collectively, our findings represent the first description of MltG function in E. faecalis and point to at least two distinct roles for MltG in PG homeostasis and cephalosporin resistance.
Importance: Enterococcus faecalis is an opportunistic pathogen that colonizes the human gut microbiome. Infections caused by E. faecalis are increasingly prevalent and difficult to treat due to the multidrug resistance exhibited toward common clinical antibiotics. A thorough understanding of the mechanisms used by E. faecalis to maintain cell wall homeostasis will serve as a foundation for future development of new therapeutics that disable enterococcal resistance to cell-wall-active antibiotics and may reveal new vulnerabilities that could be exploited by novel antimicrobials. Here, we demonstrate that the MltG peptidoglycan hydrolase is essential for enterococcal cell wall homeostasis, but that the enzymatic activity of MltG is not required for this role. Instead, the enzymatic activity of MltG impacts intrinsic resistance toward cephalosporins.
Keywords: Enterococcus; MltG; cell wall stress; cephalosporin resistance; peptidoglycan.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
-
- Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y. 2005. Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol 54:1093–1101. doi: 10.1099/jmm.0.45935-0 - DOI - PubMed
-
- Kristich C, Rice L, Arias C. 2014. Enterococcal infection-treatment and antibiotic resistance. In Gilmore M, Clewell D, Ike Y, Shankar N (ed), Enterococci: from commensals to leading causes of drug resistant infection. Massachusetts Eye and Ear Infirmary. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
