

Resolving spatial subclonal genomic heterogeneity of loss of heterozygosity and extrachromosomal DNA in gliomas
- PMID: 40514380
- PMCID: PMC12166086
- DOI: 10.1038/s41467-025-59805-z
Resolving spatial subclonal genomic heterogeneity of loss of heterozygosity and extrachromosomal DNA in gliomas
Abstract
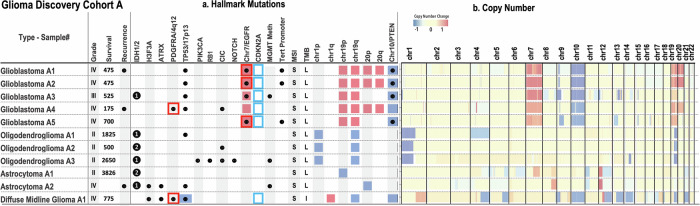
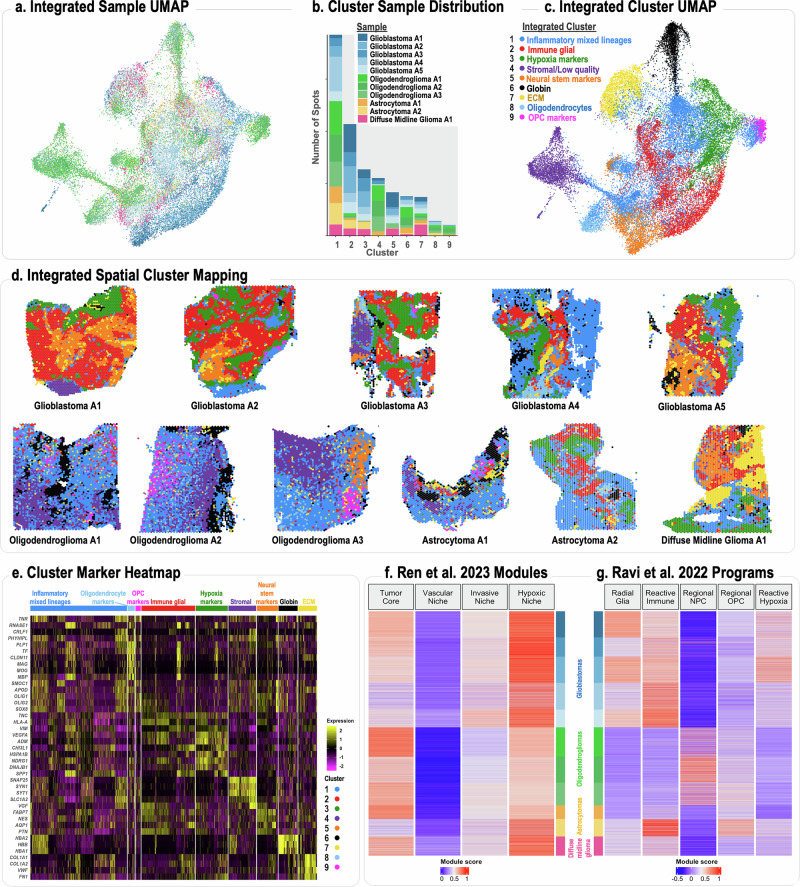
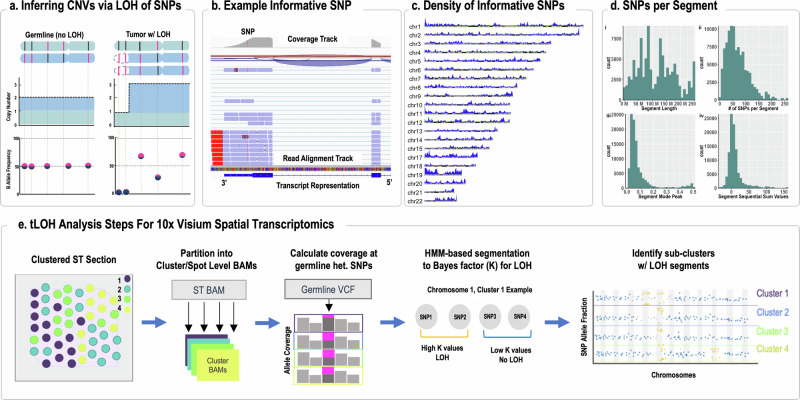
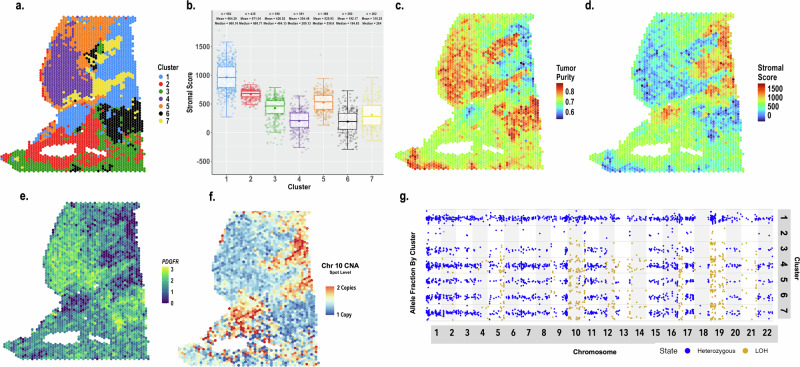
Mapping the spatial organization of DNA-level somatic copy number changes in tumors can provide insight to understanding higher-level molecular and cellular processes that drive pathogenesis. We describe an integrated framework of spatial transcriptomics, tumor/normal DNA sequencing, and bulk RNA sequencing to identify shared and distinct characteristics of an initial cohort of eleven gliomas of varied pathology and a replication cohort of six high-grade glioblastomas. We identify focally amplified extrachromosomal DNA (ecDNA) in four of the eleven initial gliomas, with subclonal tumor heterogeneity in two EGFR-amplified grade IV glioblastomas. In a TP53-mutated glioblastoma, we detect a subclone with EGFR amplification on ecDNA coupled to chromosome 17 loss of heterozygosity. To validate subclonal somatic aneuploidy and copy number alterations associated with ecDNA double minutes, we examine the replication cohort, identifying MDM2/MDM4 ecDNA subclones in two glioblastomas. The spatial heterogeneity of EGFR and p53 inactivation underscores the role of ecDNA in enabling rapid oncogene amplification and enhancing tumor adaptability under selective pressure.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
