

Augmenting precision medicine via targeted RNA-Seq detection of expressed mutations
- PMID: 40514442
- PMCID: PMC12166063
- DOI: 10.1038/s41698-025-00993-8
Augmenting precision medicine via targeted RNA-Seq detection of expressed mutations
Abstract
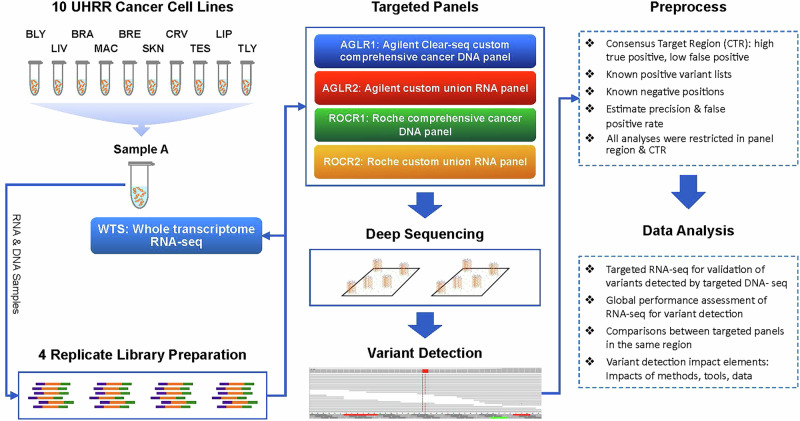
In precision medicine, DNA-based assays are currently necessary but not always sufficient for predicting therapeutic efficacy of cancer drugs based on the mutational findings in a patient's tumor specimen. Most drugs target proteins, but it is challenging and not yet cost-effective to perform high-throughput proteomics profiling, including mutational analysis, on cancer specimens. RNA may be an effective mediator for bridging the "DNA to protein divide" and provide more clarity and therapeutic predictability for precision oncology. While RNA sequencing (RNA-seq) has been increasingly used alongside DNA cancer mutation screening panels to assess the impact of variants on gene transcript expression and splicing, comprehensive evaluations of RNA panels and the integration of expressed mutation data analytics to supplement DNA panels are still limited. In this study, we conducted targeted RNA-seq on a reference sample set for expressed variant detection to explore its potential capability to complement DNA variant results or detect variants independently. The results indicated that, with a carefully controlled false positive rate ensuring high accuracy, RNA-seq uniquely identified variants with significant pathological relevance that were missed by DNA-seq, demonstrating its potential to uncover clinically actionable mutations. On the other hand, while some variants were detected by both approaches, others were missed by one or the other, reflecting either the nature of these variants or limitations of the bioinformatics tools used. Variants missed by RNA-seq are often not expressed or expressed at very low levels, suggesting they may be of lower clinical relevance. Incorporating RNA-seq into clinical biomarker panels will ultimately advance precision medicine and improve patient outcomes by improving the strength and reliability of somatic mutation findings for clinical diagnosis, prognosis and prediction of therapeutic efficacy.
© 2025. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
Competing interests: The authors declare the following potential competing financial interests: Author N.N., A.B.L., and C.P.P. were employees of Agilent Technologies, Inc., but declares no non-financial competing interests. Authors D.L., J.L., D.J.J.Jr., D.B., G.C., J.F., B.G., W.J., D.P.K, R.K., P.L., C.E.M., C.M., B.P., T.A.R., R.M., S.M.E.S., A.S., H.U.T., J.C.W., P.R.B., and J.X. declare no financial or non-financial competing interests. UHRR is a commercial product of Agilent, and sample A DNA is a potential product of Agilent Technologies, Inc.
Figures




References
LinkOut - more resources
Full Text Sources
Medical
