

Addressing the mean-variance relationship in spatially resolved transcriptomics data with spoon
- PMID: 40515599
- PMCID: PMC12166475
- DOI: 10.1093/biostatistics/kxaf012
Addressing the mean-variance relationship in spatially resolved transcriptomics data with spoon
Abstract
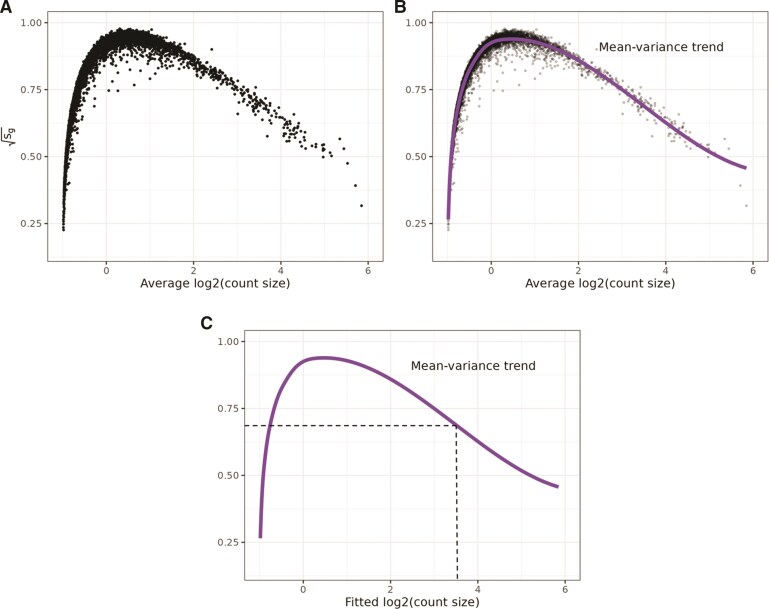
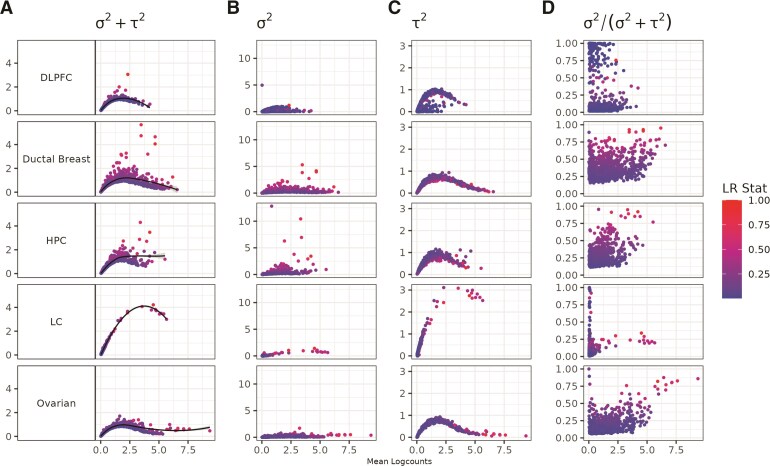
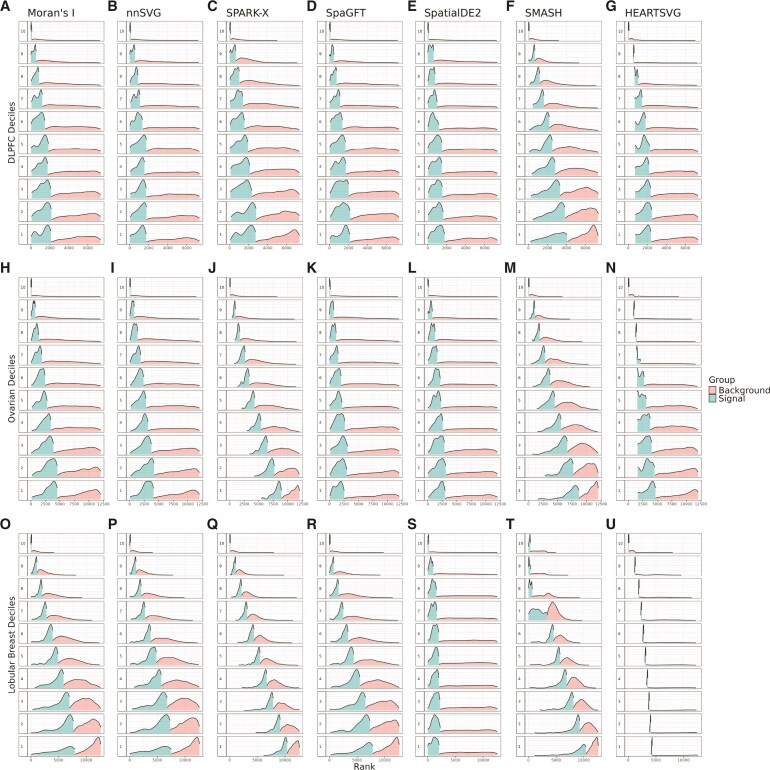
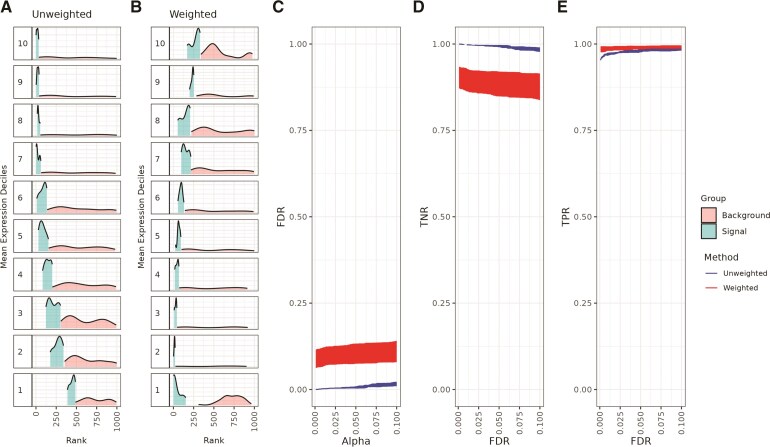
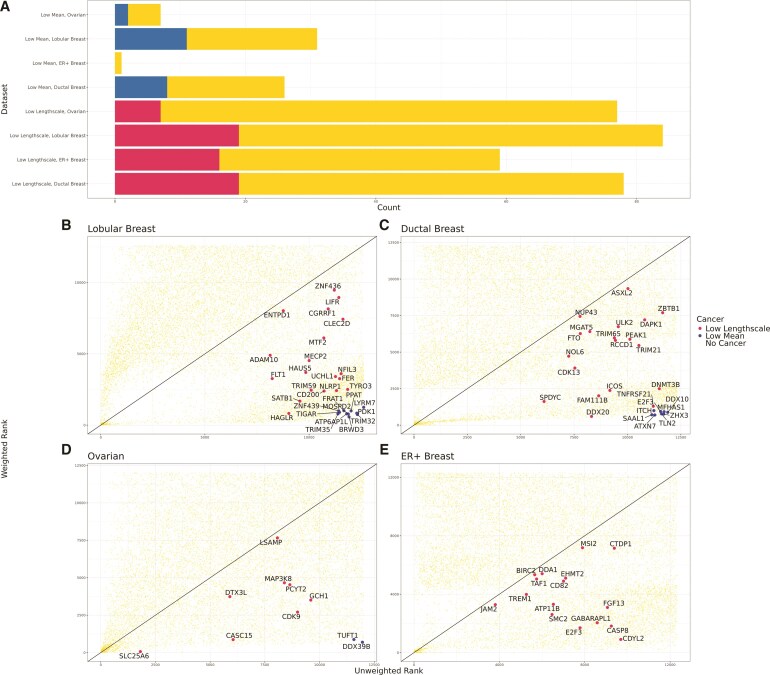
An important task in the analysis of spatially resolved transcriptomics (SRT) data is to identify spatially variable genes (SVGs), or genes that vary in a 2D space. Current approaches rank SVGs based on either $ P $-values or an effect size, such as the proportion of spatial variance. However, previous work in the analysis of RNA-sequencing data identified a technical bias with log-transformation, violating the "mean-variance relationship" of gene counts, where highly expressed genes are more likely to have a higher variance in counts but lower variance after log-transformation. Here, we demonstrate the mean-variance relationship in SRT data. Furthermore, we propose spoon, a statistical framework using empirical Bayes techniques to remove this bias, leading to more accurate prioritization of SVGs. We demonstrate the performance of spoon in both simulated and real SRT data. A software implementation of our method is available at https://bioconductor.org/packages/spoon.
Keywords: Gaussian process regression; empirical Bayes; mean–variance bias; spatial transcriptomics; spatially variable gene.
© The Author 2025. Published by Oxford University Press.
Conflict of interest statement
No competing interest is declared.
Figures
Update of
-
Addressing the mean-variance relationship in spatially resolved transcriptomics data with spoon.bioRxiv [Preprint]. 2024 Nov 8:2024.11.04.621867. doi: 10.1101/2024.11.04.621867. bioRxiv. 2024. Update in: Biostatistics. 2024 Dec 31;26(1):kxaf012. doi: 10.1093/biostatistics/kxaf012. PMID: 39574747 Free PMC article. Updated. Preprint.
References
-
- 10x Genomics. 2020. Human breast cancer: whole transcriptome analysis. https://www.10xgenomics.com/datasets/human-breast-cancer-whole-transcrip...
-
- 10x Genomics. 2022. Human breast cancer: visium fresh frozen, whole transcriptome. https://www.10xgenomics.com/resources/datasets/human-breast-cancer-visiu...
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
