

'Intelligent' proteins
- PMID: 40515853
- PMCID: PMC12167427
- DOI: 10.1007/s00018-025-05770-1
'Intelligent' proteins
Abstract
We present an idea of protein molecules that challenges the traditional view of proteins as simple molecular machines and suggests instead that they exhibit a basic form of "intelligence". The idea stems from suggestions coming from Integrated Information Theory (IIT), network theory, and allostery to explore how proteins process information, adapt to their environment, and even show memory-like behaviors. We define protein intelligence using IIT and focus on how proteins integrate information (in terms of the parameter Φ coming from IIT) and balance their core (stable, ordered regions) and periphery (flexible, disordered regions). This balance allows proteins to remain stable while adapting to changes and operating in a critical state where order and disorder coexist. We summarize recent findings on conformational memory, allosteric regulation, protein intrinsic disorder, liquid-liquid phase separation, and critical transitions, and compare protein behavior to other complex systems like ecosystems and neural networks. While our perspective offers a unified framework to understand proteins, it also raises questions about applying intelligence concepts to molecular systems. We discuss how this understanding could advance protein engineering, drug design, and synthetic biology, while at the same time acknowledging the challenges of creating adaptive, "intelligent" proteins. This concept bridges the gap between mechanistic and systems-level views of proteins and offers a comprehensive understanding of their dynamic and adaptive nature. We have tried to redefine the traditionally metaphorical concept of "intelligence" in biochemistry as a measurable property while simultaneously establishing the material foundation of protein intelligence through the identification of fundamental elements such as memory and learning in molecular systems.
Keywords: Allostery; Conformational memory; Core-periphery dynamics; Critical States; Integrated information theory; Intrinsically disordered proteins; Liquid-liquid phase separation; Post-translational modifications; Protein intelligence.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
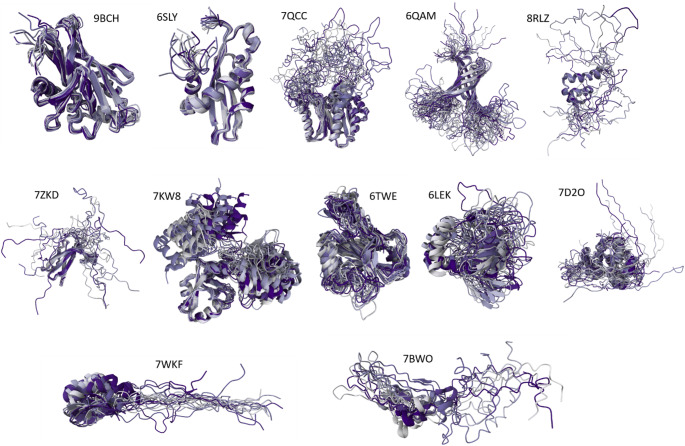
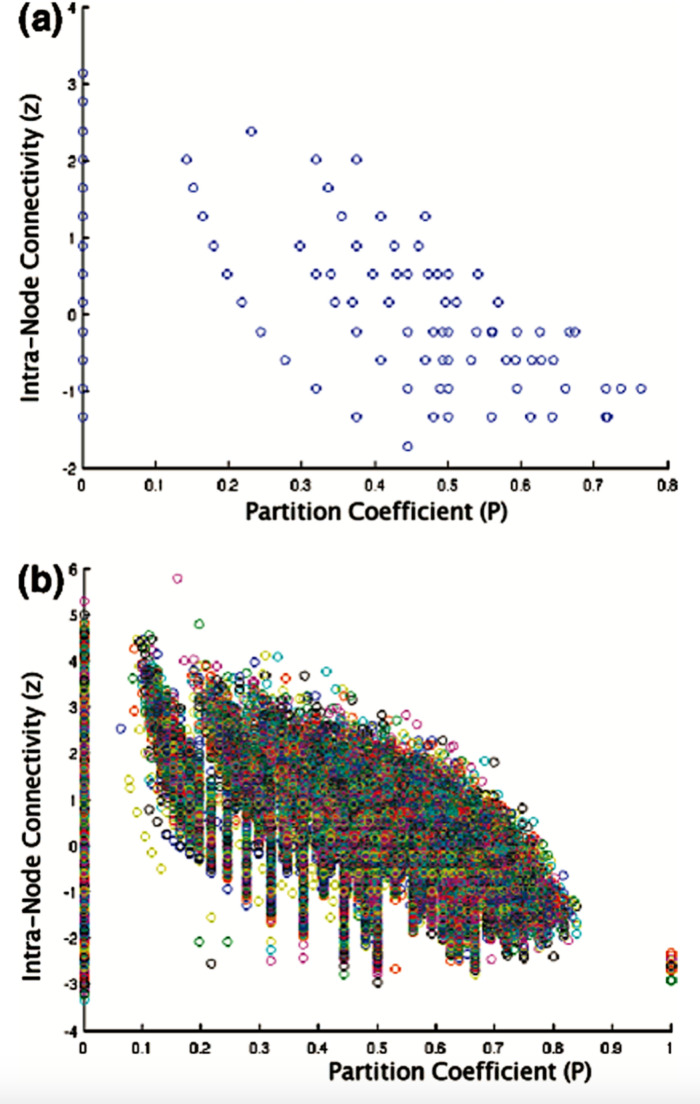
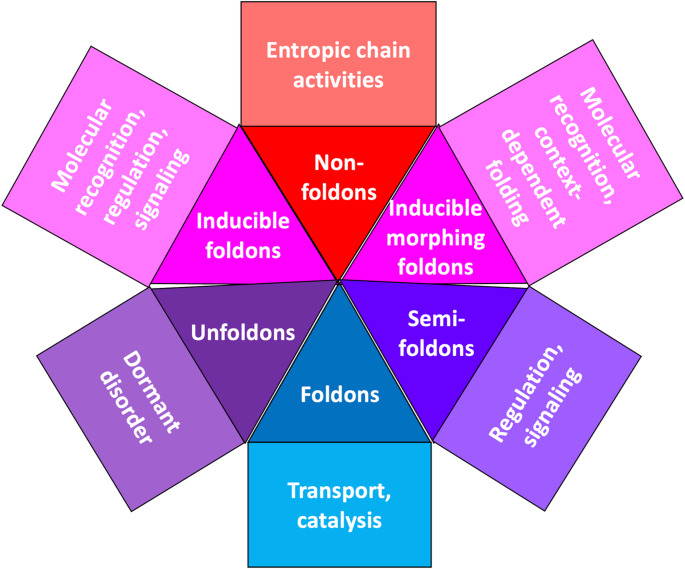
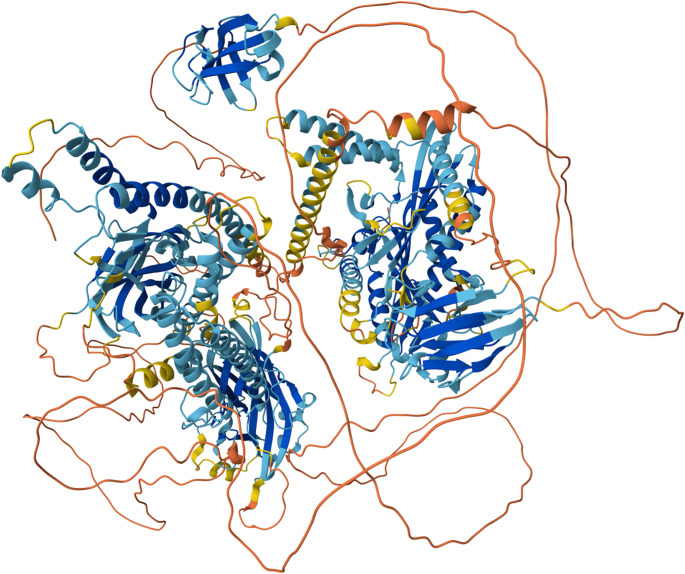
References
-
- Bruce A, Rebecca H, Alexander J, David M, Martin R, Keith R, Peter W (2022) Molecular biology of the cell: seventh international student edition with registration card. W.W. Norton & Company
-
- Schmaier AA, Zou Z, Kazlauskas A, Emert-Sedlak L, Fong KP, Neeves KB, Maloney SF, Diamond SL, Kunapuli SP, Ware J, Brass LF, Smithgall TE, Saksela K, Kahn ML (2009) Molecular priming of Lyn by GPVI enables an immune receptor to adopt a hemostatic role. Proc Natl Acad Sci U S A 106(50):21167–21172. 10.1073/pnas.0906436106 - DOI - PMC - PubMed
-
- Xu D, Phillips JC, Schulten K (1996) Protein response to external electric fields: relaxation, hysteresis, and echo. J Phys Chem 100(29). 10.1021/jp960076a
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
