

Exploring T-cell metabolism in tuberculosis: development of a diagnostic model using metabolic genes
- PMID: 40524207
- PMCID: PMC12168305
- DOI: 10.1186/s40001-025-02768-0
Exploring T-cell metabolism in tuberculosis: development of a diagnostic model using metabolic genes
Abstract
Objectives: The early diagnosis and immunoregulatory mechanisms of active tuberculosis (ATB) and latent tuberculosis infection (LTBI) remain unclear, and the role of metabolic genes in host-pathogen interactions requires further investigation.
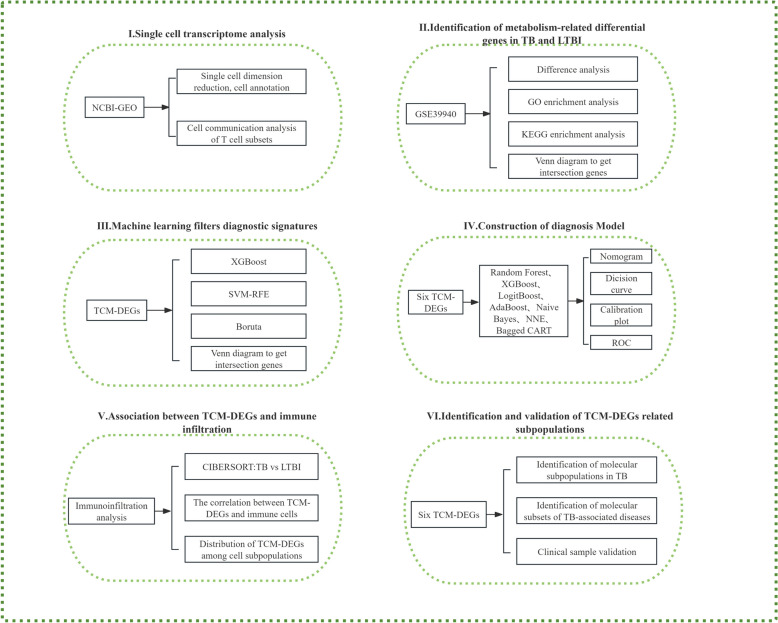
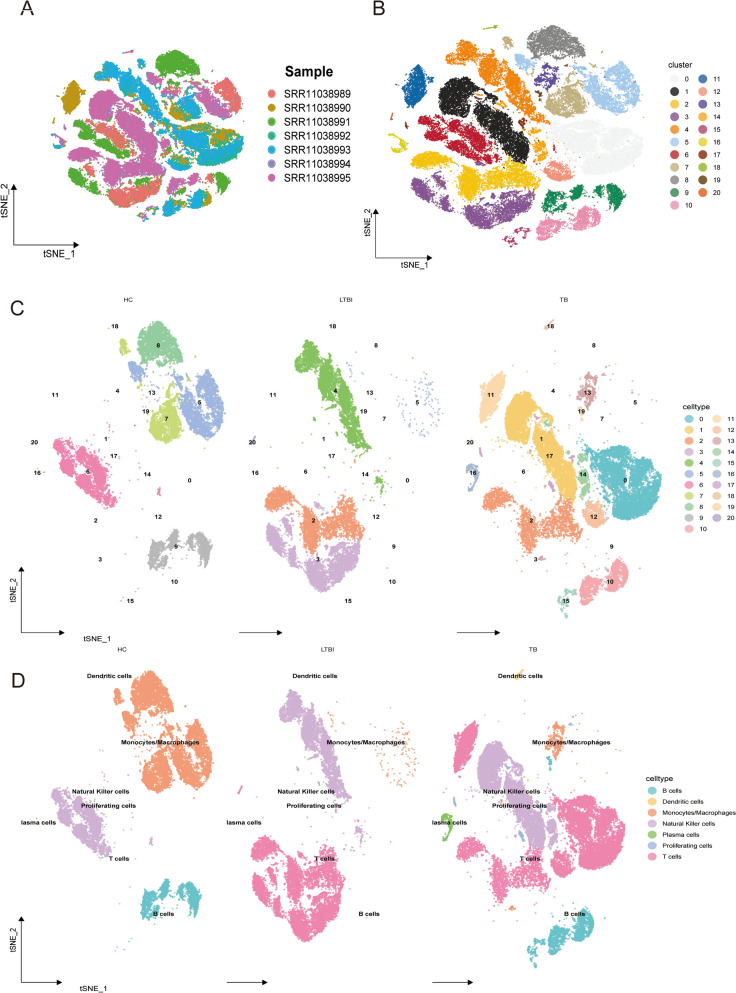
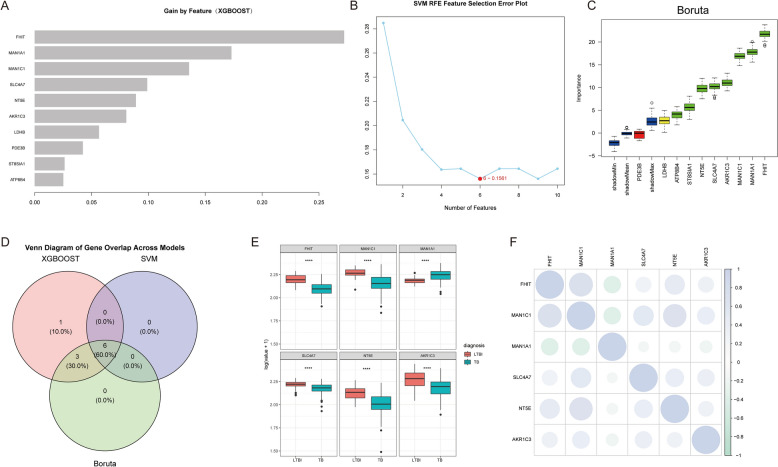
Methods: Single-cell RNA sequencing (scRNA-seq) was applied to analyze peripheral blood mononuclear cells (PBMCs) from 7 individuals, including 2 healthy controls (HC), 2 LTBI patients, and 3 ATB patients. We identified T-cell-associated metabolic differentially expressed genes (TCM-DEGs) through integrated differential expression analysis and machine learning algorithms (XGBoost, SVM-RFE, and Boruta). These TCM-DEGs were then used to construct a diagnostic model and evaluate its clinical applicability.
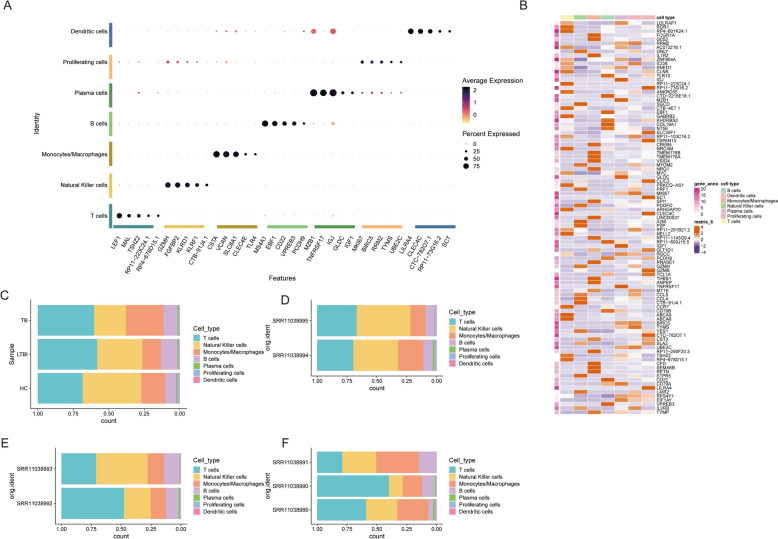
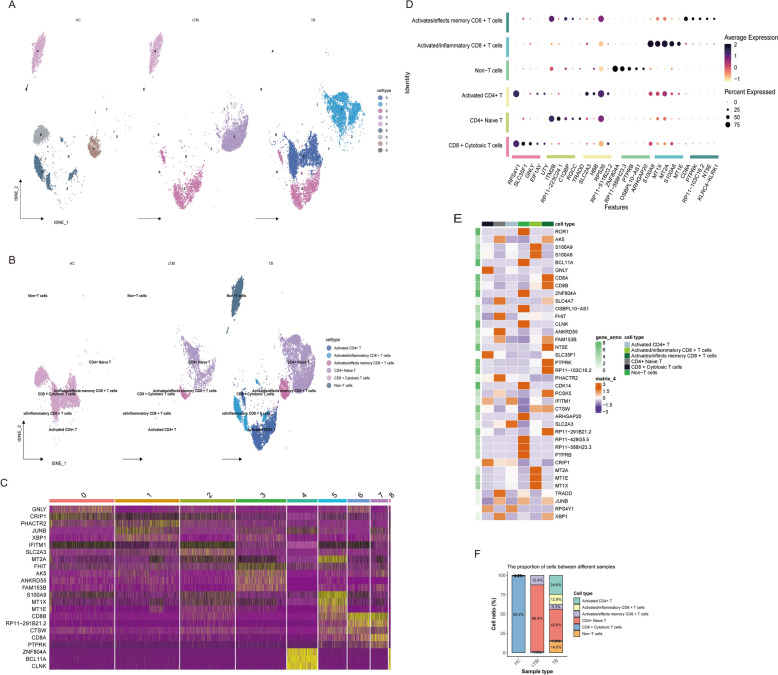
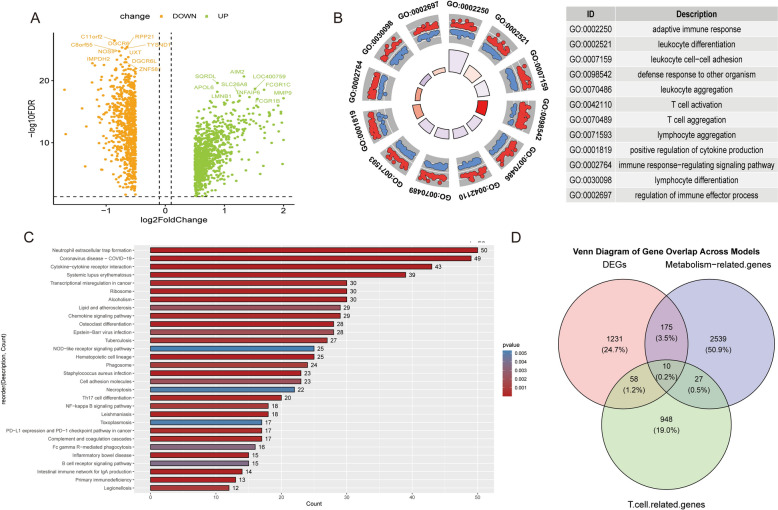
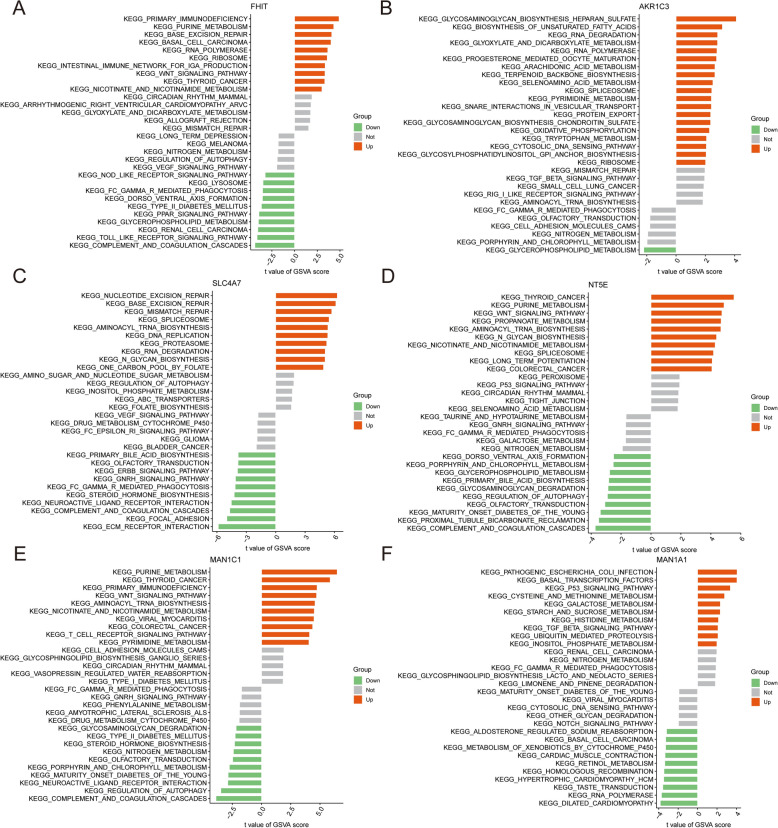
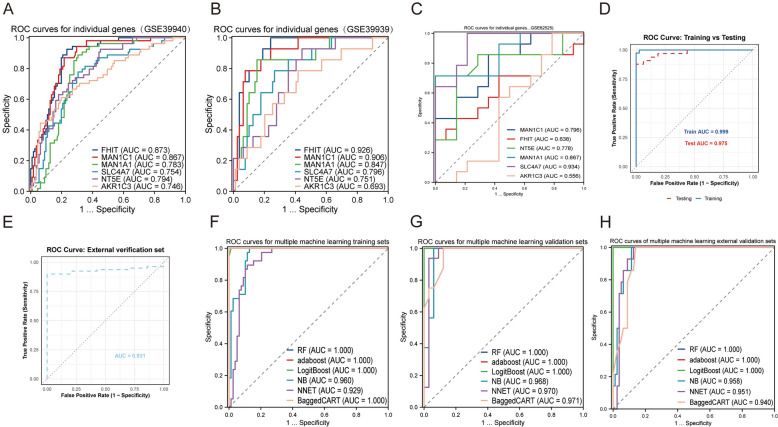
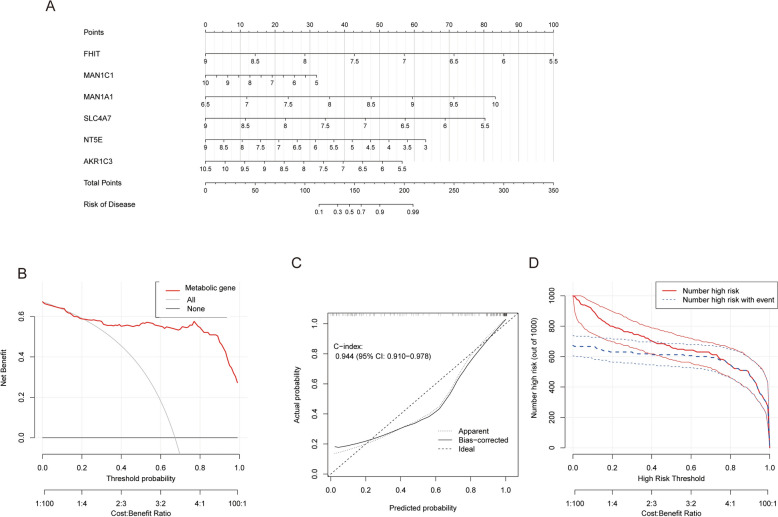
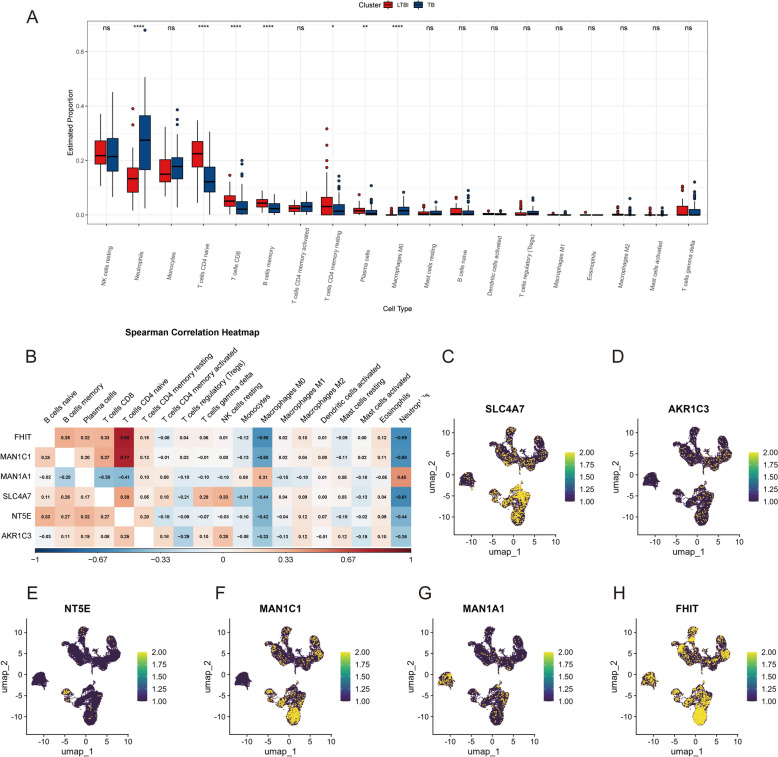
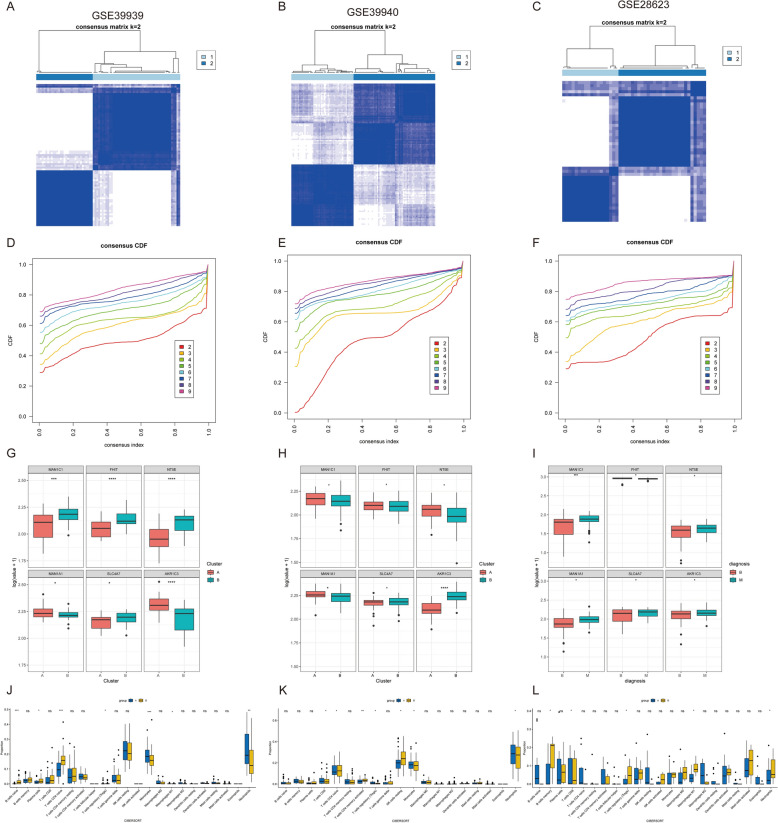
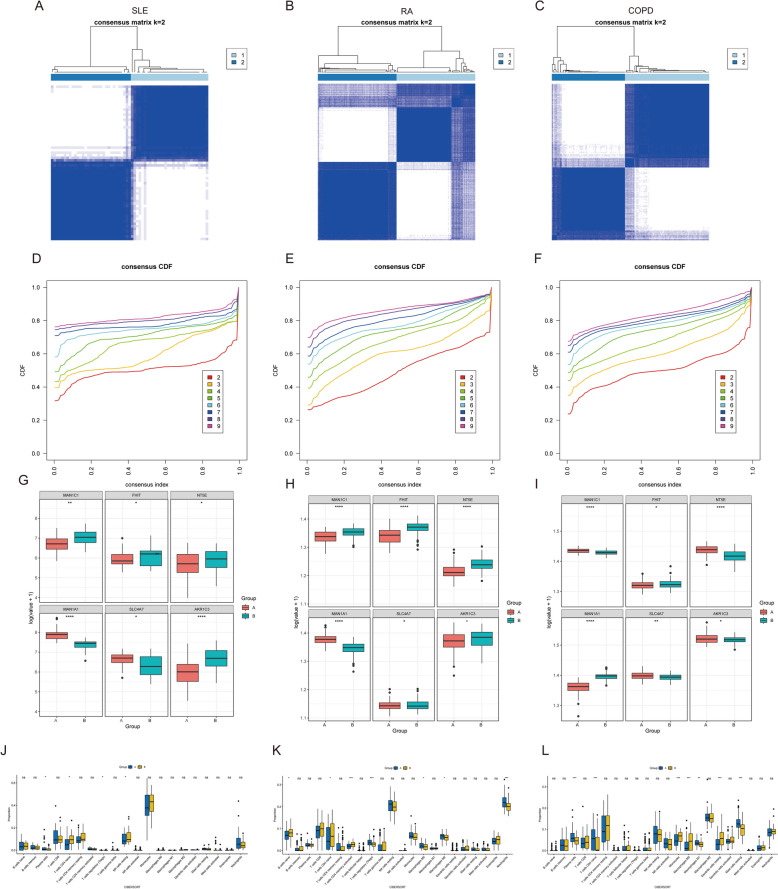
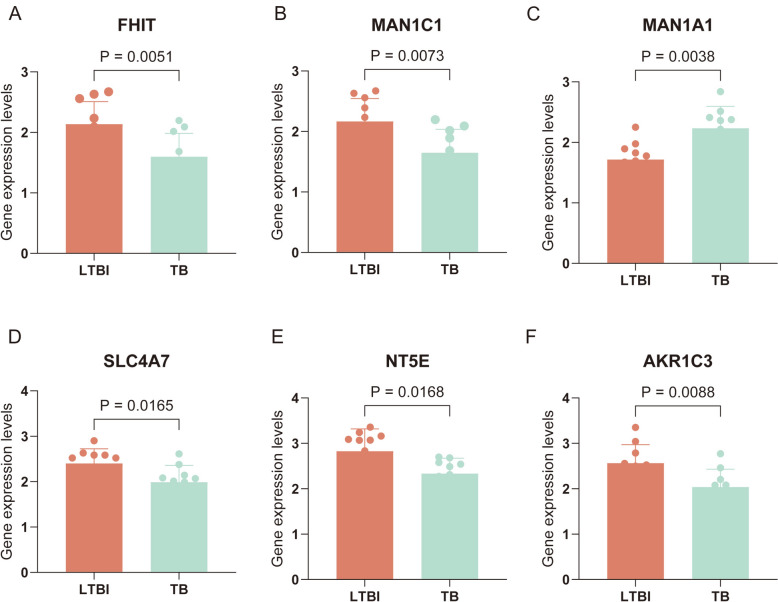
Results: The analysis revealed significant immunological alterations in TB patients, characterized by markedly elevated monocyte/macrophage populations (p < 0.001) accompanied by reduced T and NK cell counts. Notably, LTBI cases demonstrated an intermediate CD4+/CD8+ T-cell ratio, indicative of dynamic immune homeostasis. The TB cohort exhibited increased inflammatory T-cell populations, while CD8+ T-cell-mediated MHC-I and BTLA signaling pathways were identified as key regulators of immune clearance and modulation. Transcriptomic profiling identified five metabolically significant differentially expressed genes (FHIT, MAN1C1, SLC4C7, NT5E, AKR1C3; p < 0.05) that effectively distinguish between latent tuberculosis infection (LTBI) and active tuberculosis (TB). The machine learning-driven diagnostic framework demonstrated remarkable consistency across independent validation cohorts (GSE39940, GSE39939), exhibiting AUC values spanning 0.867-0.873. Molecular subtyping analysis delineated two distinct TB phenotypes: an immune-activated M1 macrophage-dominant subtype and a CD8 + T-cell infiltrated immunophenotype. Clinical validation substantiated the differential expression patterns of T-cell-related metabolic differentially expressed genes (TCM-DEGs; p < 0.05), while the nomogram predictive model achieved exceptional discriminative capacity (C-index = 0.944), demonstrating superior clinical applicability through decision curve analysis.
Conclusions: Our findings reveal that TCM-DEGs critically regulate TB progression through immune-metabolic reprogramming and cell-cell communication networks. The developed diagnostic model and molecular subtyping strategy enable precise TB-LTBI differentiation and inform immunotherapy optimization.
Keywords: Machine learning biomarkers; Metabolic gene signatures; Molecular subtypes; T-cell metabolism; Tuberculosis.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: This study has been approved by the Ethics Committee of Siyang Hospital with the approval number HY2024005. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures

Similar articles
-
Identification of cuproptosis-related molecular subtypes as a biomarker for differentiating active from latent tuberculosis in children.BMC Genomics. 2023 Jul 1;24(1):368. doi: 10.1186/s12864-023-09491-2. BMC Genomics. 2023. PMID: 37393262 Free PMC article.
-
Transcriptomic and proteomic signatures of host NK cells delineate distinct immune states across tuberculosis infection statuses.Front Immunol. 2025 Jun 16;16:1607770. doi: 10.3389/fimmu.2025.1607770. eCollection 2025. Front Immunol. 2025. PMID: 40589756 Free PMC article.
-
Autophagy-related molecular clusters identified as indicators for distinguishing active and latent TB infection in pediatric patients.BMC Pediatr. 2024 Jun 19;24(1):398. doi: 10.1186/s12887-024-04881-1. BMC Pediatr. 2024. PMID: 38890657 Free PMC article.
-
Host-response-based gene signatures for tuberculosis diagnosis: A systematic comparison of 16 signatures.PLoS Med. 2019 Apr 23;16(4):e1002786. doi: 10.1371/journal.pmed.1002786. eCollection 2019 Apr. PLoS Med. 2019. PMID: 31013272 Free PMC article.
-
From immunology to artificial intelligence: revolutionizing latent tuberculosis infection diagnosis with machine learning.Mil Med Res. 2023 Nov 28;10(1):58. doi: 10.1186/s40779-023-00490-8. Mil Med Res. 2023. PMID: 38017571 Free PMC article. Review.
References
-
- Trajman A, Campbell JR, Kunor T, et al. Tuberculosis. Lancet. 2025;405(10481):850–66. - PubMed
-
- Krutikov M, Faust L, Nikolayevskyy V, et al. The diagnostic performance of novel skin-based in-vivo tests for tuberculosis infection compared with purified protein derivative tuberculin skin tests and blood-based in vitro interferon-gamma release assays: a systematic review and meta-analysis. Lancet Infect Dis. 2022;22(2):250–64. - PubMed
-
- Jonas DE, Riley SR, Lee LC, et al. Screening for latent tuberculosis infection in adults: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2023;329(17):1495–509. - PubMed
-
- Jaccard A, Wyss T, Maldonado-Pérez N, et al. Reductive carboxylation epigenetically instructs T cell differentiation. Nature. 2023;621(7980):849–56. - PubMed
MeSH terms
Grants and funding
- 2024SY005/Siyang Hospital and the Affiliated Hospital of Jiangsu University in the first session of the hospital science and technology programmed projects to set up the topic
- 2024SY005/Siyang Hospital and the Affiliated Hospital of Jiangsu University in the first session of the hospital science and technology programmed projects to set up the topic
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
