

Multidimensional bioinformatics analysis of chondrosarcoma subtypes and TGF-β signaling networks using big data approaches
- PMID: 40526169
- PMCID: PMC12174002
- DOI: 10.1007/s12672-025-02931-3
Multidimensional bioinformatics analysis of chondrosarcoma subtypes and TGF-β signaling networks using big data approaches
Abstract
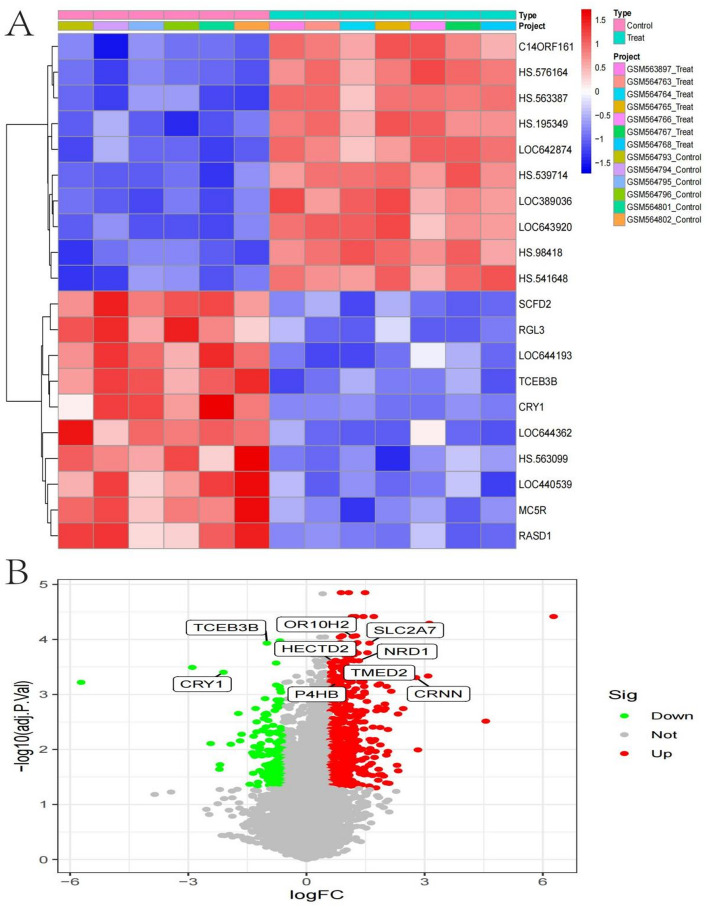
Background: Chondrosarcoma, a rare and heterogeneous malignant bone tumor, presents significant clinical challenges due to its complex molecular underpinnings and limited treatment options. In this study, we employ single-cell RNA sequencing (scRNA-seq) and bioinformatics analyses to delineate cell subtypes, decipher signaling networks, and identify gene expression patterns, thereby providing novel insights into potential therapeutic targets and their implications in cancer biology.
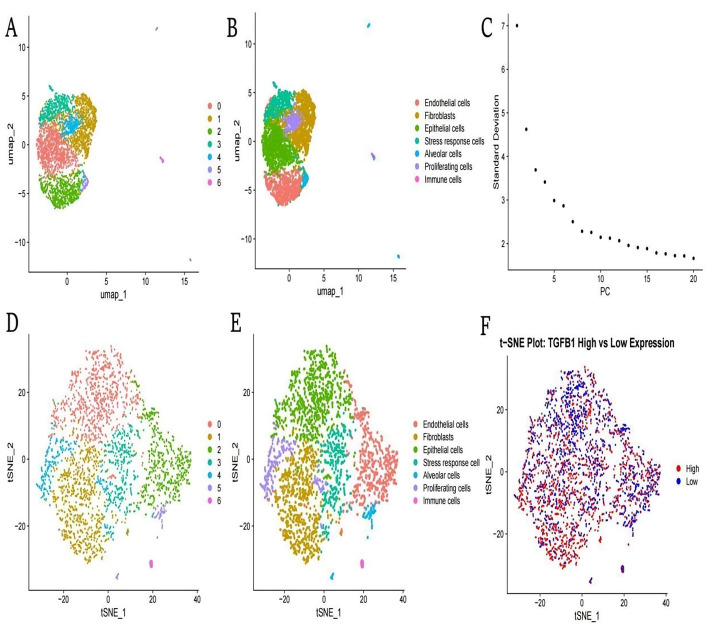
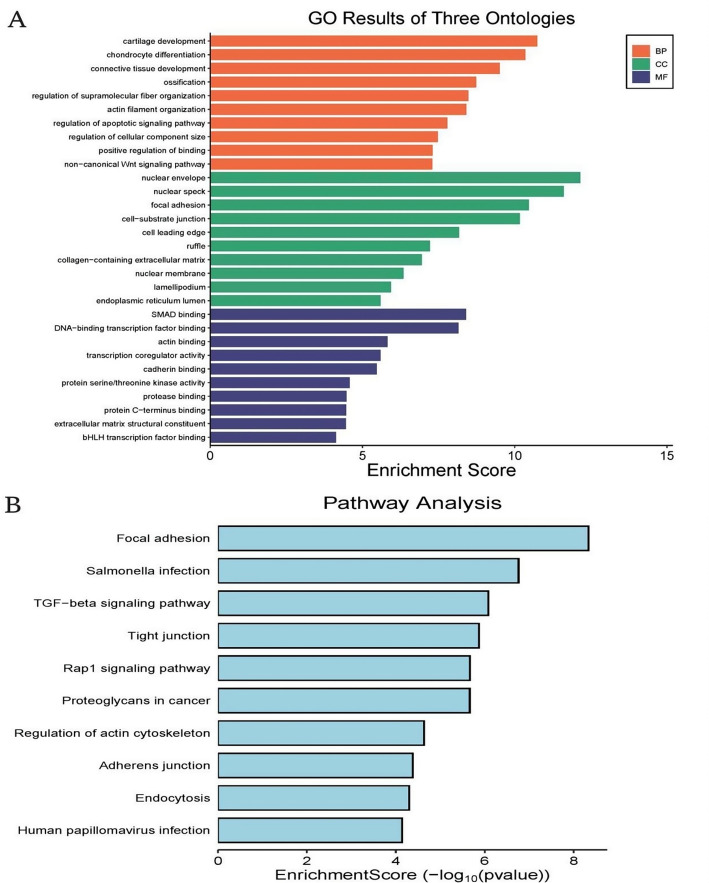
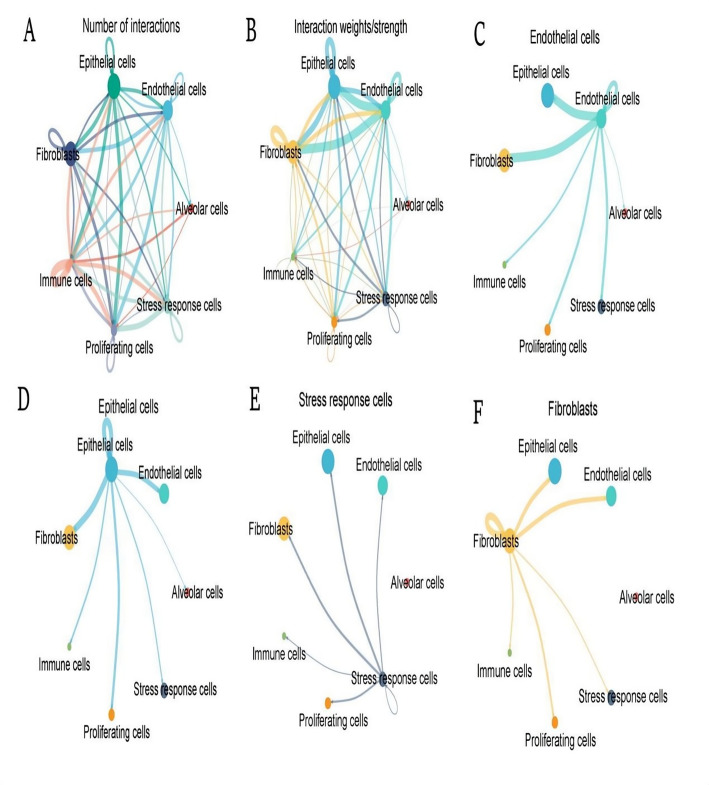
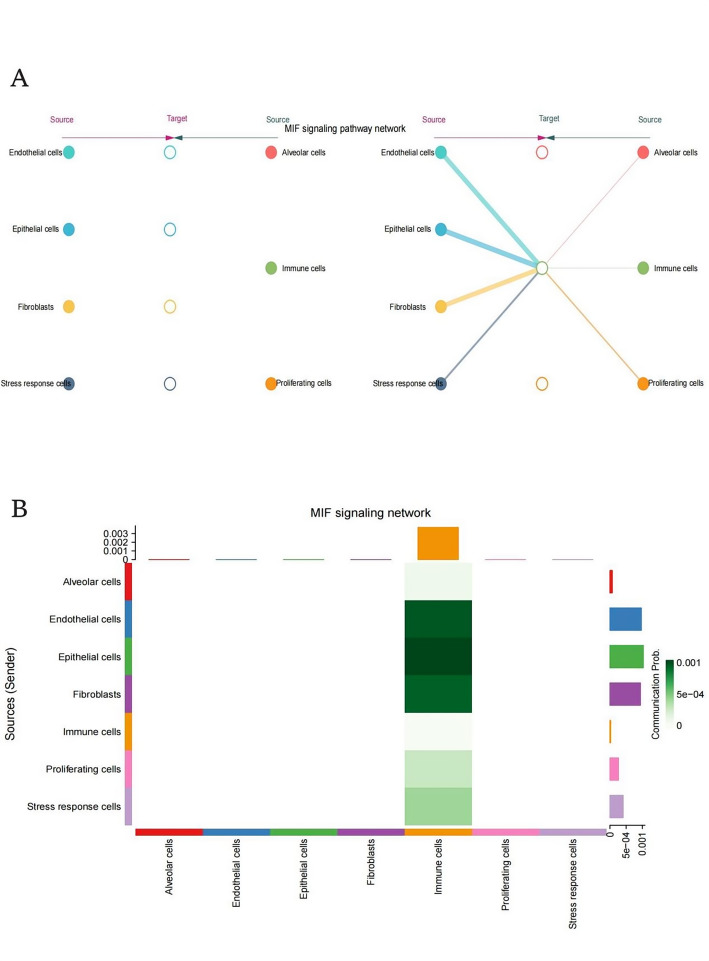
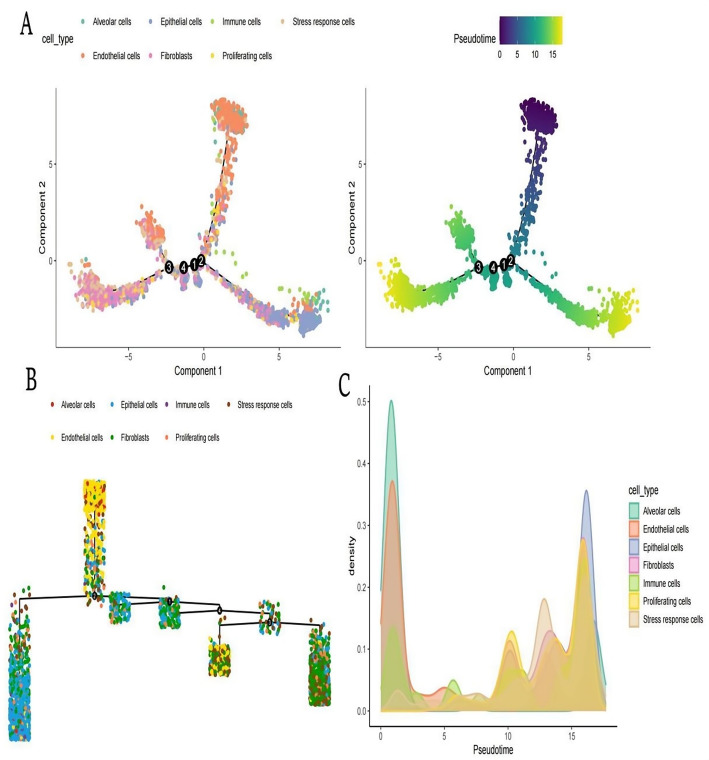
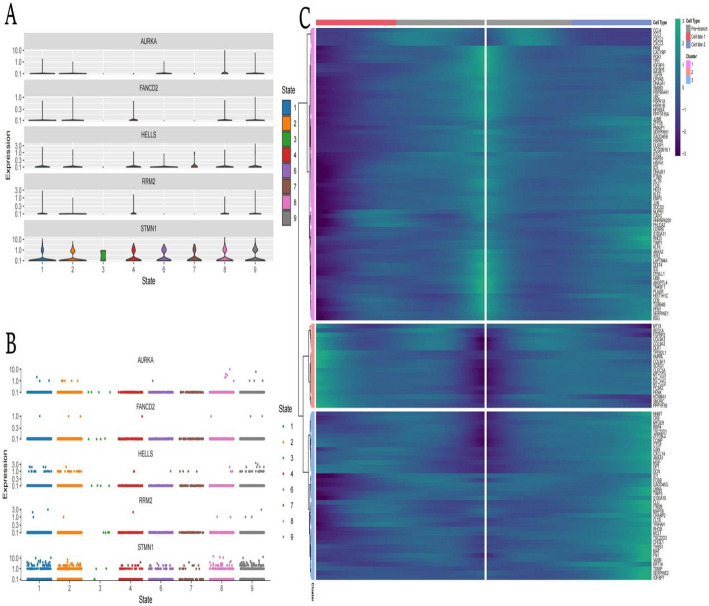
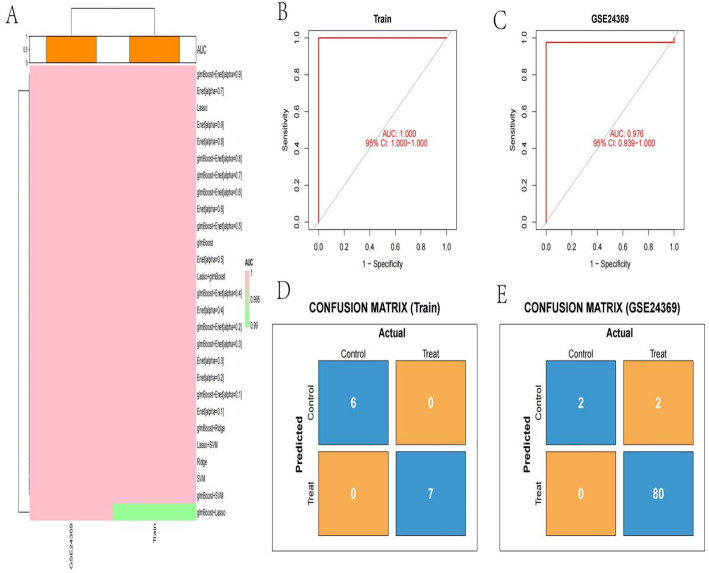
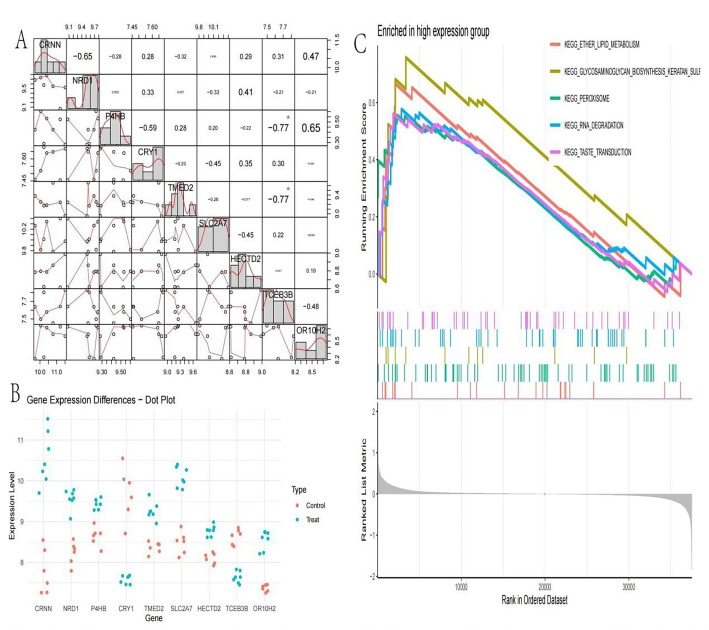
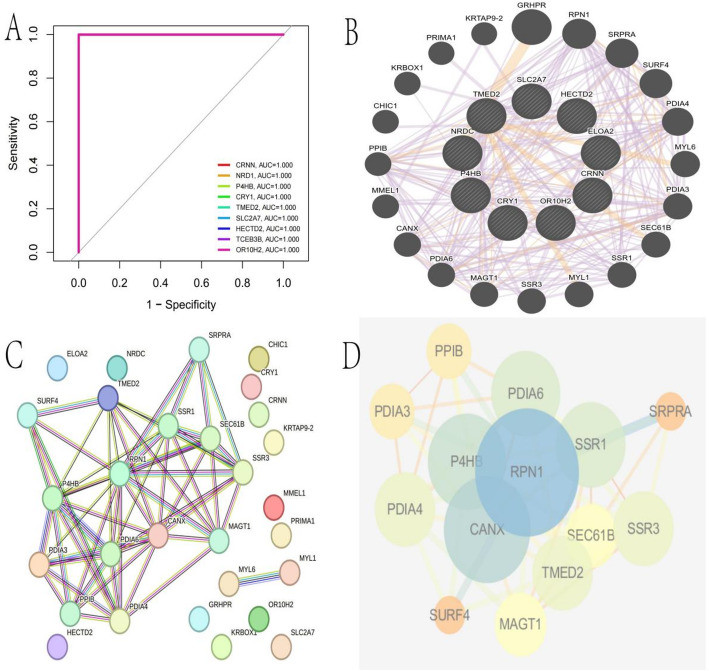
Methods: scRNA-seq was performed on both clinical and experimental chondrosarcoma samples. Dimensionality reduction techniques (UMAP/t-SNE) were used to cluster cell subtypes, followed by Gene Ontology (GO) and pathway analyses to elucidate their biological functions. Cell-cell interaction networks, including the MIF signaling network, were reconstructed to map intercellular communications. Pseudotime analysis charted differentiation trajectories, while machine learning models evaluated the classification accuracy of gene expression patterns. GSEA was conducted to identify state-specific differential expression profiles.
Results: Over ten distinct cell subtypes were identified, including endothelial cells, fibroblasts, and epithelial cells. Key signaling pathways, such as TGF-beta signaling, focal adhesion, and actin cytoskeleton regulation, were found to mediate intercellular interactions. The MIF signaling network underscored the critical roles of immune cells within the tumor microenvironment. Pseudotime analysis revealed dynamic differentiation states, while state-specific gene expression patterns emerged from GSEA. Machine learning models demonstrated robust classification performance across training and external validation datasets.
Conclusions: This comprehensive analysis uncovers the cellular heterogeneity and complex intercellular networks in chondrosarcoma, elucidating critical molecular pathways and identifying novel therapeutic targets. By integrating gene expression, signaling networks, and advanced computational methods, this study contributes to the broader understanding of cancer biology and highlights the potential for precision medicine strategies in treating chondrosarcoma.
Keywords: Bioinformatics analysis; Cell subtypes; Chondrosarcoma; Single-cell RNA sequencing; TGF-beta signaling.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: This study did not involve any human participants or animal subjects, and therefore ethical approval is not applicable. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Molecular feature-based classification of retroperitoneal liposarcoma: a prospective cohort study.Elife. 2025 May 23;14:RP100887. doi: 10.7554/eLife.100887. Elife. 2025. PMID: 40407808 Free PMC article.
-
Integrated machine learning and single-cell RNA sequencing reveal COL4A2 and CXCL6 as oxidative stress-associated biomarkers in periodontitis.Front Immunol. 2025 Jun 5;16:1598642. doi: 10.3389/fimmu.2025.1598642. eCollection 2025. Front Immunol. 2025. PMID: 40539067 Free PMC article.
-
Pathway-based cancer transcriptome deciphers a high-resolution intrinsic heterogeneity within bladder cancer classification.J Transl Med. 2025 Jun 17;23(1):666. doi: 10.1186/s12967-025-06682-1. J Transl Med. 2025. PMID: 40528211 Free PMC article.
-
A bibliometric analysis of research trends in mesenchymal stem cell therapy for neonatal bronchopulmonary dysplasia: 2004-2024.Front Pediatr. 2025 Jun 3;13:1558301. doi: 10.3389/fped.2025.1558301. eCollection 2025. Front Pediatr. 2025. PMID: 40530182 Free PMC article. Review.
-
A meta-analysis of genome-wide association studies to identify candidate genes associated with feed efficiency traits in pigs.J Anim Sci. 2025 Jan 4;103:skaf010. doi: 10.1093/jas/skaf010. J Anim Sci. 2025. PMID: 39847436 Free PMC article.
References
-
- Al-Mourgi M, Shams A. A rare entity of the anterior chest cage rib chondrosarcoma: a case report and review of literature. Semin Thorac Cardiovasc Surg. 2024. 10.1053/j.semtcvs.2024.09.005. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous