

Integrated multi-omics analysis and predictive modeling of heart failure using sepsis-related gene signature
- PMID: 40526611
- PMCID: PMC12173235
- DOI: 10.1371/journal.pone.0326212
Integrated multi-omics analysis and predictive modeling of heart failure using sepsis-related gene signature
Abstract
Background: Heart failure (HF) is characterized by complex molecular alterations, and recent studies suggest a potential role for sepsis-related genes in cardiovascular dysfunction. This study aimed to develop a predictive model for HF based on sepsis-related gene signatures.
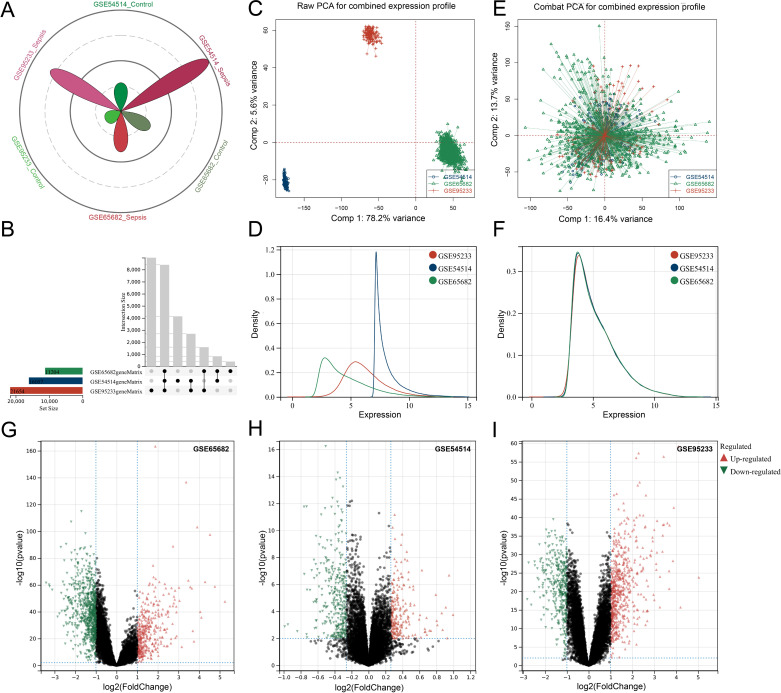
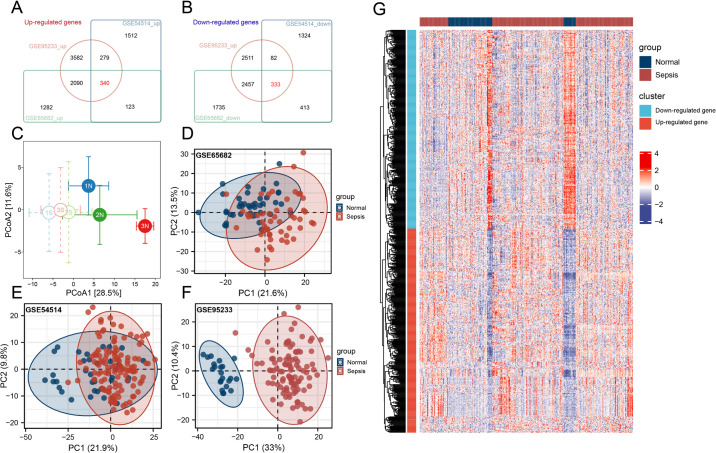
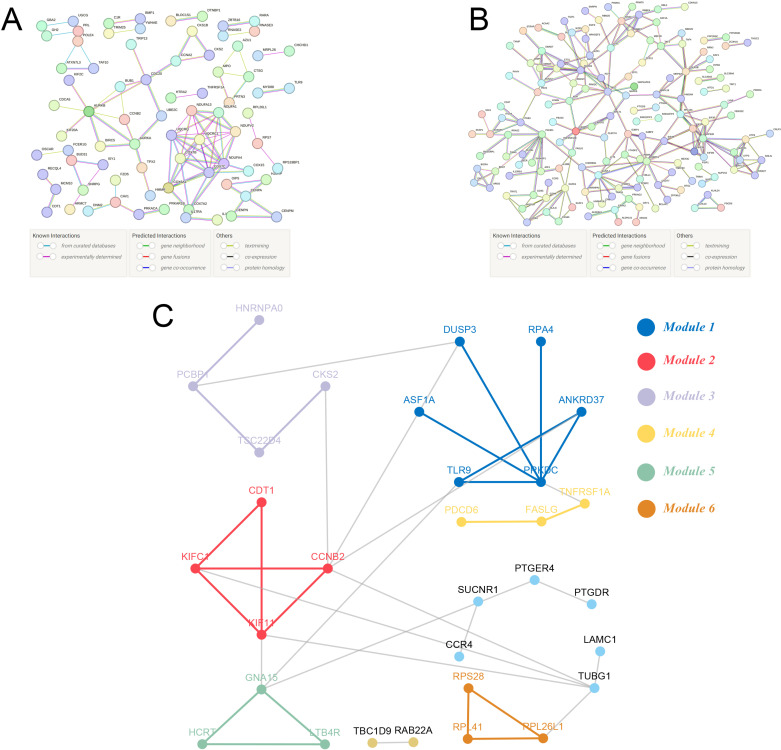
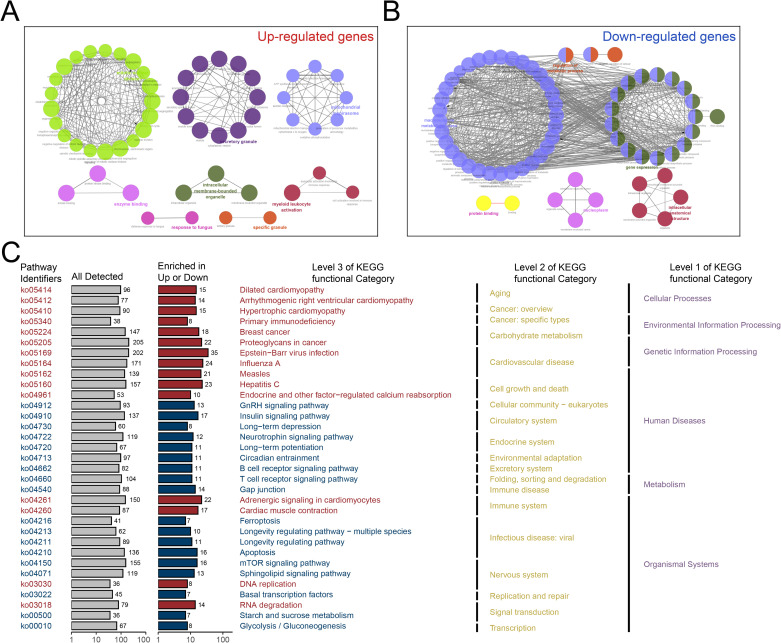
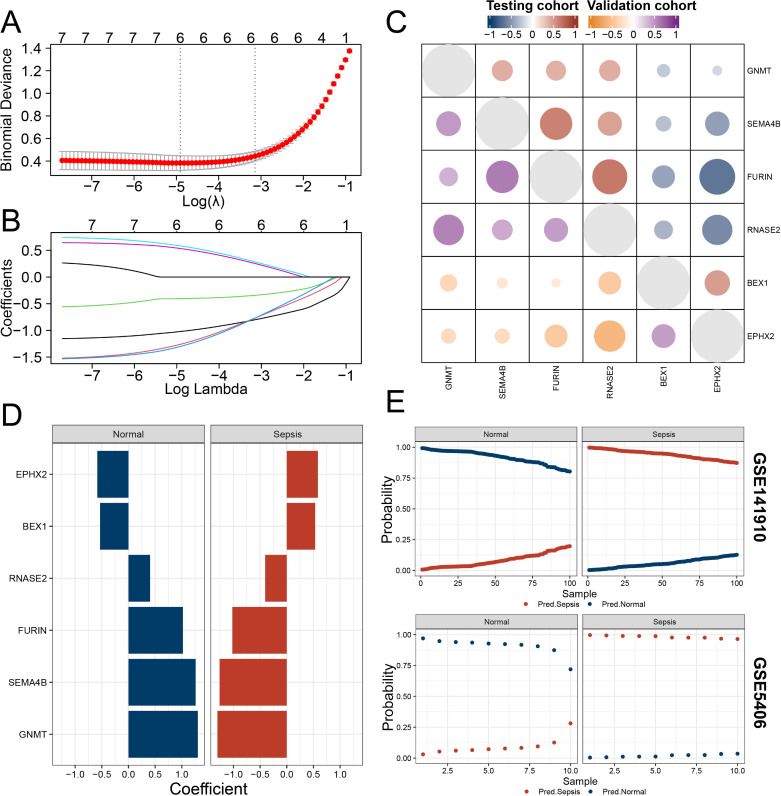
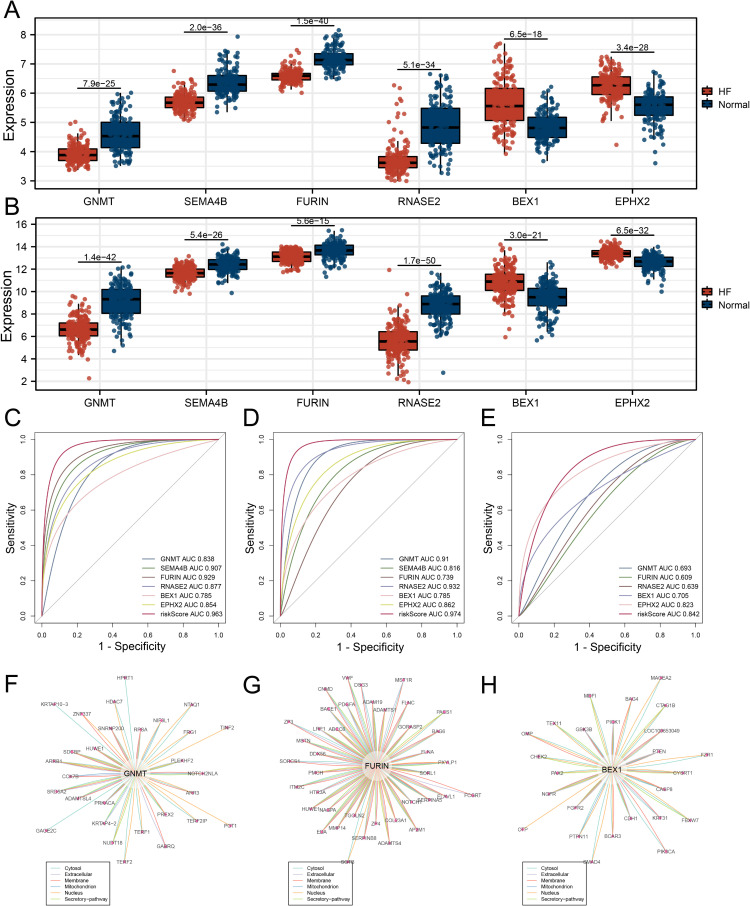
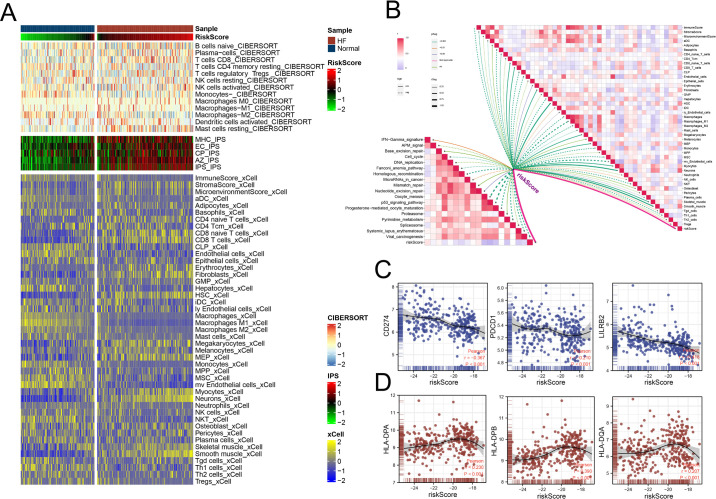
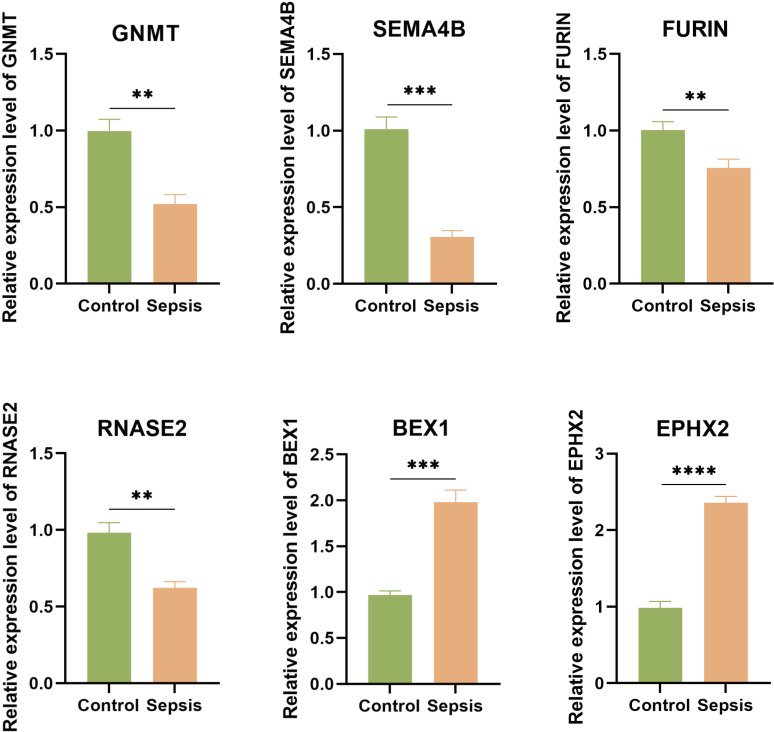
Methods: Three sepsis-related datasets (GSE65682, GSE54514, and GSE95233) were analyzed to identify differentially expressed genes (DEGs) following batch effect correction using the ComBat algorithm. With the use of elastic net regularization and the glmnet package in R, Lasso Cox regression was employed to screen out gene signatures. A predictive model was developed based on the expression of each gene signature and the co-efficient values. In addition, the predictive model was validated on independent HF datasets (GSE57345, GSE141910, and GSE5406). Model performance was assessed through receiver operating characteristic (ROC) analysis and AUC values of each gene signature, and immune infiltration was evaluated using CIBERSORT, IPS, and xCell. Sepsis models of C57BL/6 mice were established by cecal ligation and puncture (CLP).
Results: We identified 340 up-regulated and 333 down-regulated sepsis-related genes. The predictive model, incorporating six key genes, demonstrated superior performance compared to individual genes across both training and validation datasets with the AUC value of the risk score above 0.9, significantly higher than that of a single gene. Immune infiltration profiles differed significantly between HF patients and controls, with more pronounced alterations observed at higher risk score levels. Finally, the expression of six key genes in sepsis models was confirmed to be consistent with our prediction.
Conclusion: The model constructed through sepsis-related characteristic genes provides a highly advantageous method for predicting HF, and the characteristic genes we have screened may be potential biomarkers for predicting HF. This model has potential application value in early diagnosis and risk stratification, which can help improve the clinical management of heart failure and provide new ideas for preventing HF.
Copyright: © 2025 Lang et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Developing a Panel of Shared Susceptibility Genes as Diagnostic Biomarkers for chronic obstructive pulmonary disease and Heart Failure.Comput Biol Med. 2025 Sep;196(Pt A):110657. doi: 10.1016/j.compbiomed.2025.110657. Epub 2025 Jul 4. Comput Biol Med. 2025. PMID: 40617086
-
Molecular feature-based classification of retroperitoneal liposarcoma: a prospective cohort study.Elife. 2025 May 23;14:RP100887. doi: 10.7554/eLife.100887. Elife. 2025. PMID: 40407808 Free PMC article.
-
Multi-omics Analysis Reveals Molecular Subtype of Mitochondrial Oxidative Stress and Prognostic Model Development in Sepsis.Front Biosci (Landmark Ed). 2025 Jun 20;30(6):38957. doi: 10.31083/FBL38957. Front Biosci (Landmark Ed). 2025. PMID: 40613299
-
Assessing the comparative effects of interventions in COPD: a tutorial on network meta-analysis for clinicians.Respir Res. 2024 Dec 21;25(1):438. doi: 10.1186/s12931-024-03056-x. Respir Res. 2024. PMID: 39709425 Free PMC article. Review.
-
Defining disease severity in atopic dermatitis and psoriasis for the application to biomarker research: an interdisciplinary perspective.Br J Dermatol. 2024 Jun 20;191(1):14-23. doi: 10.1093/bjd/ljae080. Br J Dermatol. 2024. PMID: 38419411 Free PMC article. Review.
References
-
- Shankar-Hari M, Harrison DA, Rubenfeld GD. Epidemiology of sepsis and septic shock in critical care units: comparison between sepsis-2 and sepsis-3 populations using a national critical care database. British Journal of Anaesthesia. 2017;119(4):626–36. - PubMed
-
- Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ. 2016;353:i1585. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
