

Mumemto: efficient maximal matching across pangenomes
- PMID: 40528225
- PMCID: PMC12172372
- DOI: 10.1186/s13059-025-03644-0
Mumemto: efficient maximal matching across pangenomes
Abstract
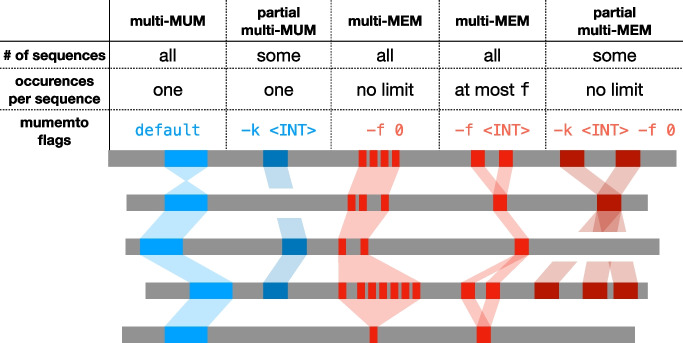
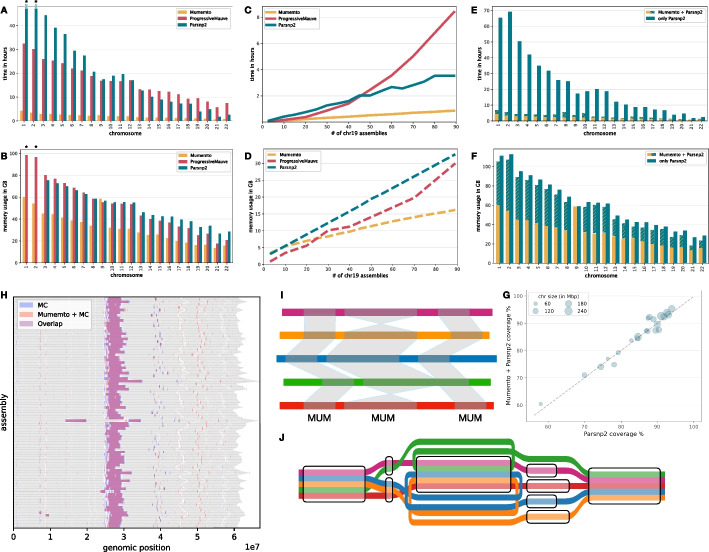
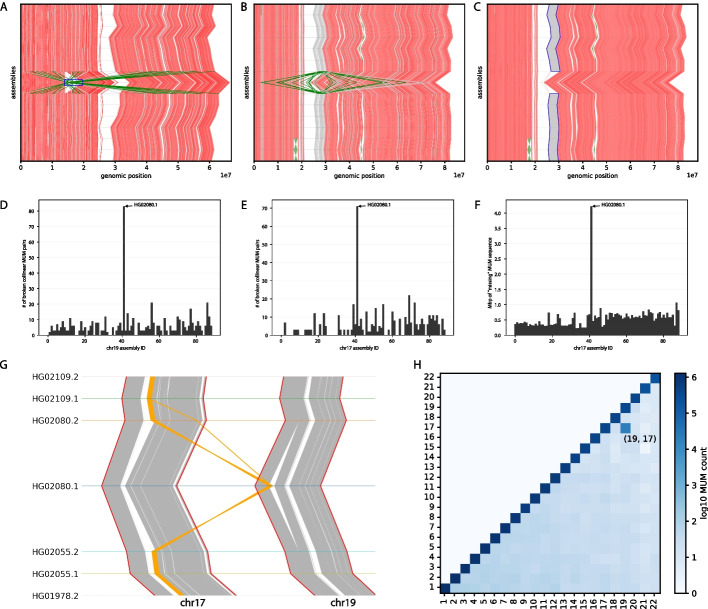
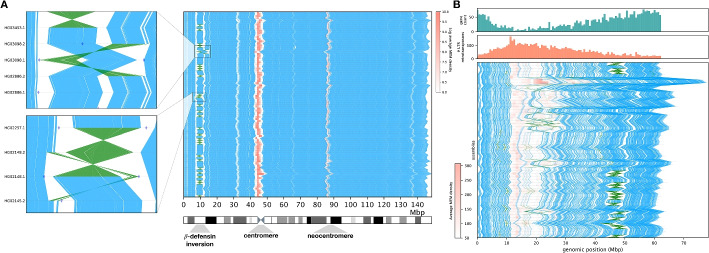
Aligning genomes into common coordinates is central to pangenome construction, though computationally expensive. Multi-sequence maximal unique matches (multi-MUMs) help to frame and solve the multiple alignment problem. We introduce Mumemto, a tool that computes multi-MUMs and other match types across large pangenomes. Mumemto allows for visualization of synteny, reveals aberrant assemblies and scaffolds, and highlights pangenome conservation and structural variation. Mumemto computes multi-MUMs across 320 human assemblies (960GB) in 25.7 h with 800 GB of memory and hundreds of fungal assemblies in minutes. Mumemto is implemented in C++ and Python and available open-source at https://github.com/vikshiv/mumemto (v1.1.1 at doi.org/10.5281/zenodo.15053447 ).
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethical approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures

Update of
-
Mumemto: efficient maximal matching across pangenomes.bioRxiv [Preprint]. 2025 Jan 5:2025.01.05.631388. doi: 10.1101/2025.01.05.631388. bioRxiv. 2025. Update in: Genome Biol. 2025 Jun 17;26(1):169. doi: 10.1186/s13059-025-03644-0. PMID: 39803467 Free PMC article. Updated. Preprint.
References
-
- Abouelhoda MI, Kurtz S, Ohlebusch E. Replacing suffix trees with enhanced suffix arrays. J Discret Algoritm. 2004;2(1):53–86.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
