

Ensemble Docking for Intrinsically Disordered Proteins
- PMID: 40532196
- PMCID: PMC12264940
- DOI: 10.1021/acs.jcim.5c00370
Ensemble Docking for Intrinsically Disordered Proteins
Abstract
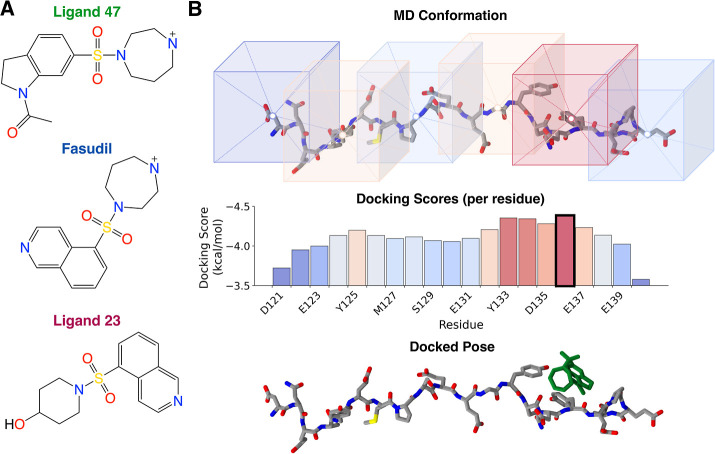
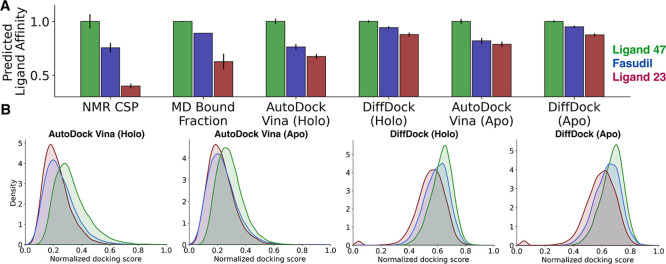
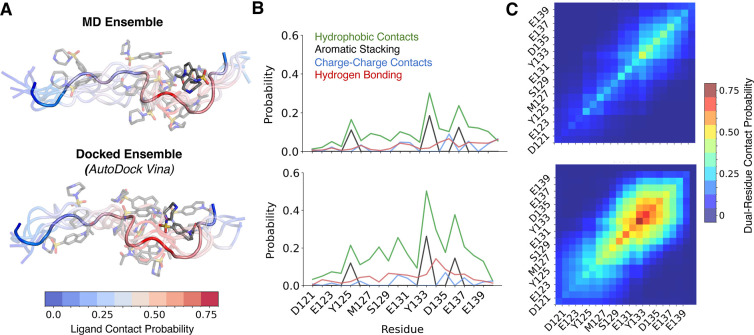
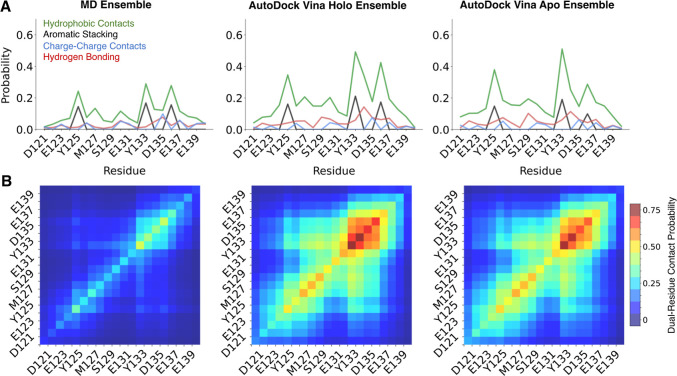
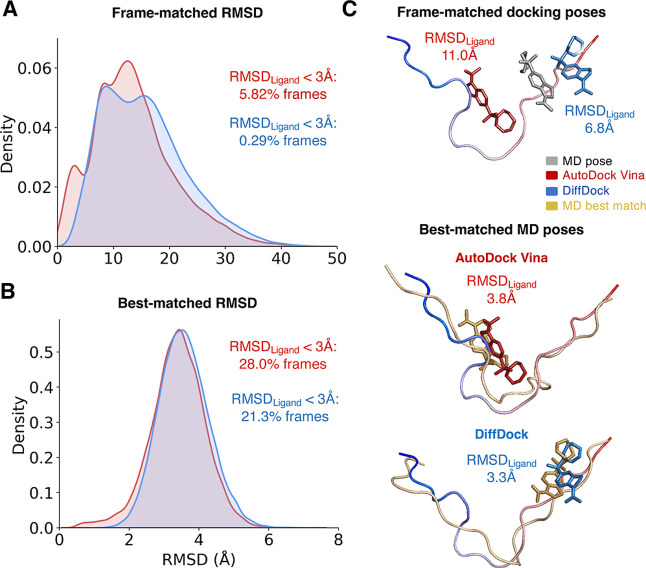
Intrinsically disordered proteins (IDPs) are implicated in many human diseases and are increasingly being pursued as drug targets. Conventional structure-based drug design methods that rely on well-defined binding sites are, however, largely unsuitable for IDPs. Here, we present computationally efficient ensemble docking approaches to predict the relative affinities of small molecules to IDPs and characterize their dynamic, heterogeneous binding mechanisms at atomic resolution. We show that these ensemble docking protocols accurately predict the relative binding affinities of three small molecule α-synuclein ligands measured by NMR spectroscopy and generate conformational ensembles of ligand binding modes in remarkable agreement with experimentally validated long-time scale molecular dynamics simulations of ligand binding. Our results demonstrate the potential of ensemble docking approaches for predicting small molecule binding to IDPs and suggest that these methods may be valuable tools for IDP drug discovery campaigns.
Figures
Update of
-
Ensemble docking for intrinsically disordered proteins.bioRxiv [Preprint]. 2025 Jan 26:2025.01.23.634614. doi: 10.1101/2025.01.23.634614. bioRxiv. 2025. Update in: J Chem Inf Model. 2025 Jul 14;65(13):6847-6860. doi: 10.1021/acs.jcim.5c00370. PMID: 39896485 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
