

Exploring the genetic diversity of the 3' untranslated region (UTR) of Dengue virus
- PMID: 40533782
- PMCID: PMC12178008
- DOI: 10.1186/s12985-025-02800-z
Exploring the genetic diversity of the 3' untranslated region (UTR) of Dengue virus
Abstract
Background: Dengue virus (DENV), with its four antigenically distinct serotypes, is the etiological agent of dengue fever, which is endemic in tropical and subtropical regions but has recently spread to previously non-endemic areas such as Europe. Given the growing body of evidence suggesting that sequence diversification in the 3' UTR of DENV contributes to its epidemiological fitness and host adaptation, we conducted phylogenetic and genetic diversity analyses on four DENV 3' UTR datasets (DENV-1, DENV-2, DENV-3 and DENV-4).
Methods: A maximum likelihood phylogenetic analysis was performed using IQ-TREE, and the average evolutionary divergence was estimated using MEGA X. Maximum likelihood analysis combined with genetic distance calculations provided insight into the evolutionary dynamics of the DENV 3' UTR.
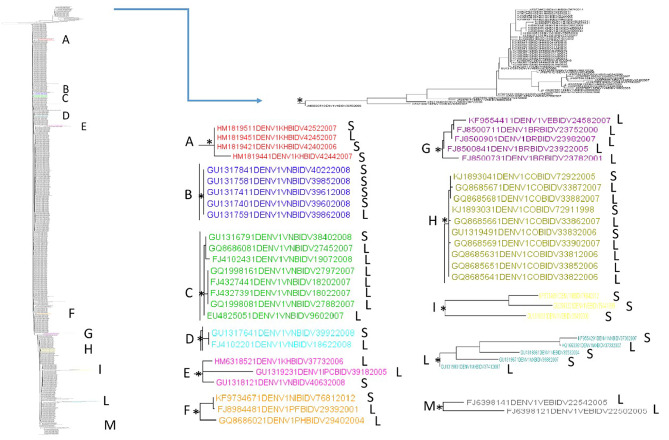
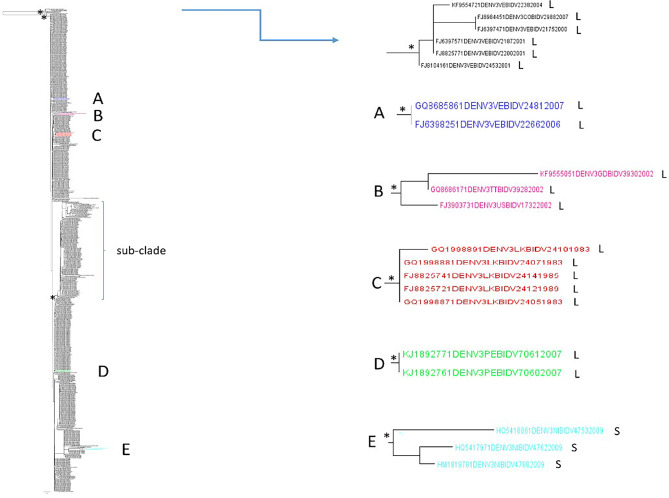
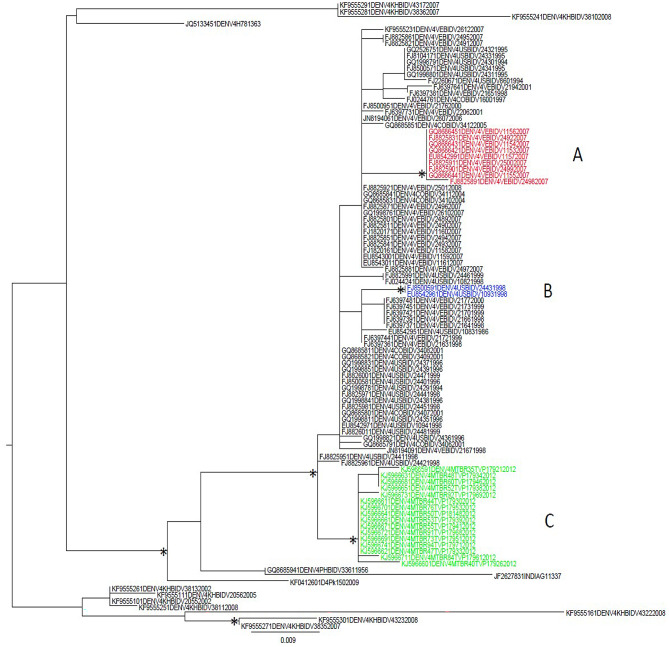
Results: A higher number of supported internal clusters/clades were found in the DENV-2 and DENV-1 3' UTR trees, probably indicating strains with similar evolutionary histories. In terms of cluster composition, apart from a general mixing of DENV 3' UTR sequences from different sites and years, the majority of supported internal clusters were composed of sequences aggregated according to their location. Genetic distances showed that the DENV-1 3' UTR has a higher variability (5%) compared to DENV-2 (3%), DENV-3 and DENV-4 (2%). The average length of the 3' UTR, as estimated from our datasets, showed that it was longest in DENV-2, followed by DENV-3, DENV-1 and DENV-4.
Conclusions: In conclusion, this study provided a comprehensive analysis of 3' UTR evolution and phylogenies in all four DENV serotypes, suggesting that this viral genomic sequence is subject to genetic variability and length changes.
Keywords: 3′ UTR; DENV; Dengue virus; Evolution; Genetic diversity; Phylogenesis.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures
References
-
- Monath TP. The arboviruses: epidemiology and ecology. Boca Raton, FL, USA: CRC; 2021. Volume 5.
-
- Frasca F, Sorrentino L, Fracella M, D’Auria A, Coratti E, Maddaloni L, Bugani G, Gentile M, Pierangeli A, d’Ettorre G, et al. An update on the entomology, virology, pathogenesis, and epidemiology status of West nile and dengue viruses in Europe (2018–2023). Trop Med Infect Disease. 2024;9:166. 10.3390/tropicalmed9070166. - PMC - PubMed
-
- Naddaf M. Mosquito-borne diseases are surging in Europe — how worried are scientists? Nature. 2024;633:749–749. 10.1038/d41586-024-03031-y. - PubMed
-
- Naddaf M. Dengue is spreading in Europe: how worried should we be? Nature. 2023. 10.1038/d41586-023-03407-6. - PubMed
Publication types
MeSH terms
Substances
Associated data
- BioProject/31235
- Actions
Grants and funding
- Project no. PE00000007, INF-ACT/EU funding within the NextGenerationEU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases
- Project no. PE00000007, INF-ACT/EU funding within the NextGenerationEU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases
- (Anna Tramontano, Call 2024)./grant from Istituto Pasteur Italia-Fondazione Cenci Bolognetti
LinkOut - more resources
Full Text Sources
