

Integrated transcriptomics and machine learning reveal REN as a dual regulator of tumor stemness and NK cell evasion in Wilms tumor progression
- PMID: 40534868
- PMCID: PMC12174124
- DOI: 10.3389/fimmu.2025.1612987
Integrated transcriptomics and machine learning reveal REN as a dual regulator of tumor stemness and NK cell evasion in Wilms tumor progression
Abstract
Introduction: Wilms tumor (WT) is the most common pediatric kidney cancer, which presents significant therapeutic challenges, particularly in high-risk cases, due to chemotherapy resistance and immunosuppressive tumor microenvironments (TMEs). Tumor stemness and immune evasion mechanisms are implicated in poor clinical outcomes, yet the molecular drivers underpinning these processes remain inadequately understood.
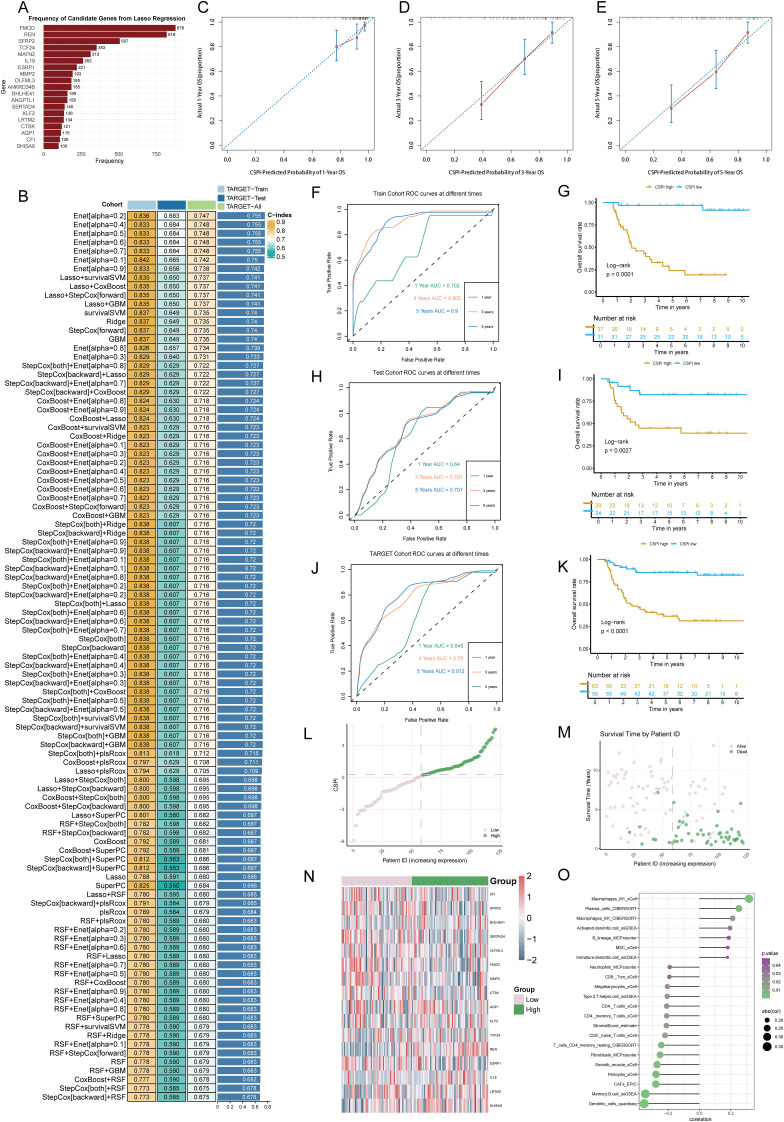
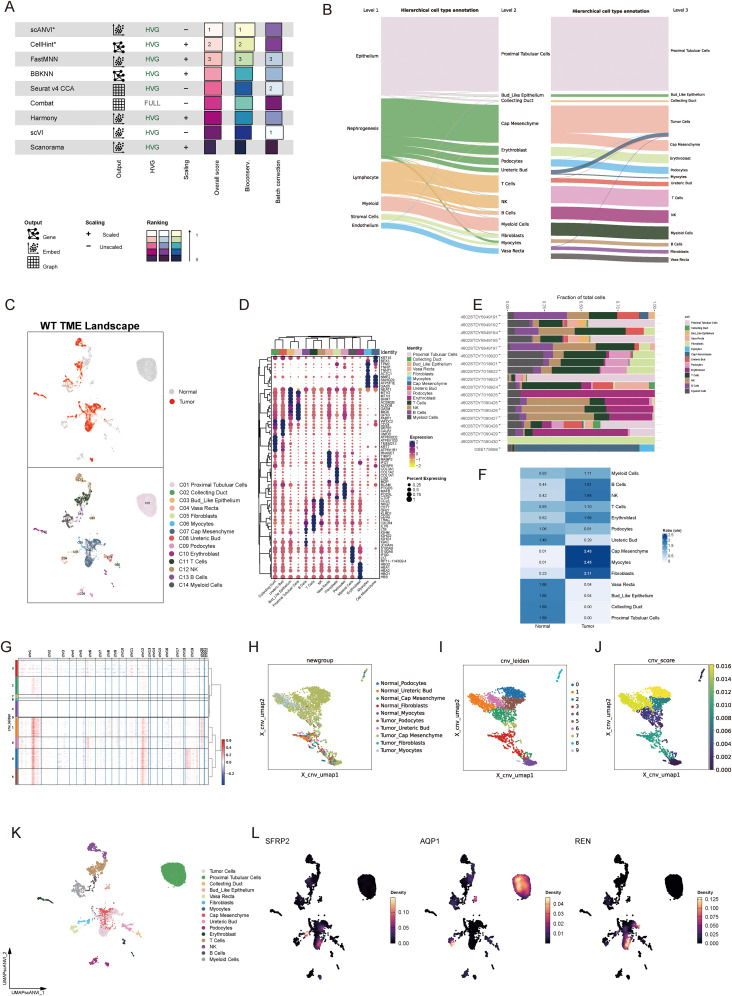
Methods: We employed an integrative approach combining single-cell RNA sequencing (scRNA-seq), spatial transcriptomics, bulk RNA-seq, and advanced machine learning techniques to uncover molecular regulators of tumor behavior in WT. A novel Cancer Stemness Prognostic Index (CSPI) was developed using machine learning algorithms to stratify WT patients by risk and histological subtype. Additionally, molecular docking simulations and in vitro functional assays were performed to validate the role of key regulators in tumor stemness and immune evasion, as well as to explore potential therapeutic strategies targeting these molecular drivers.
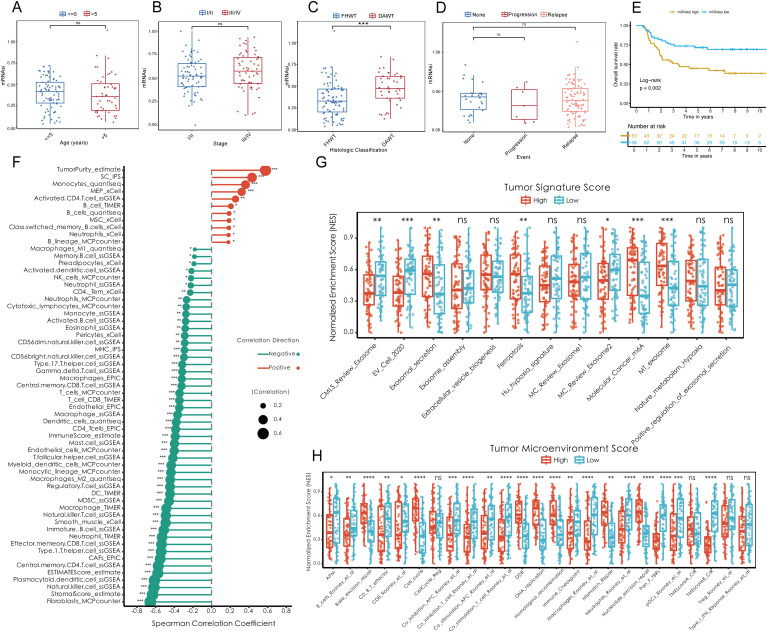
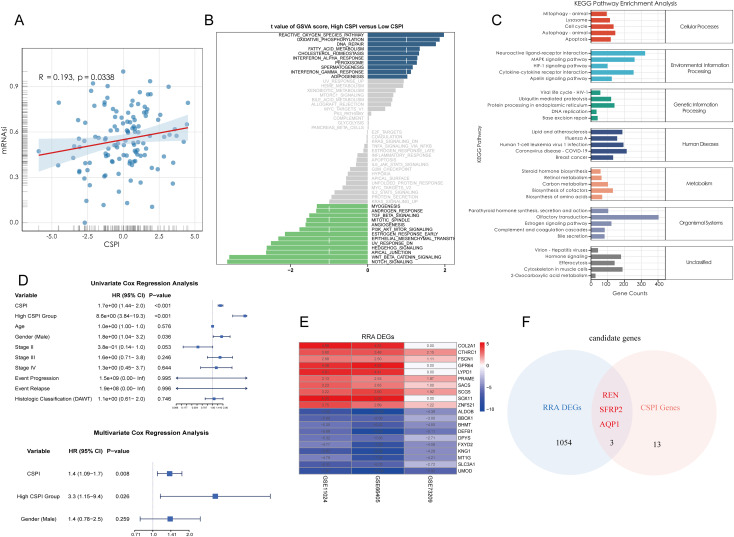
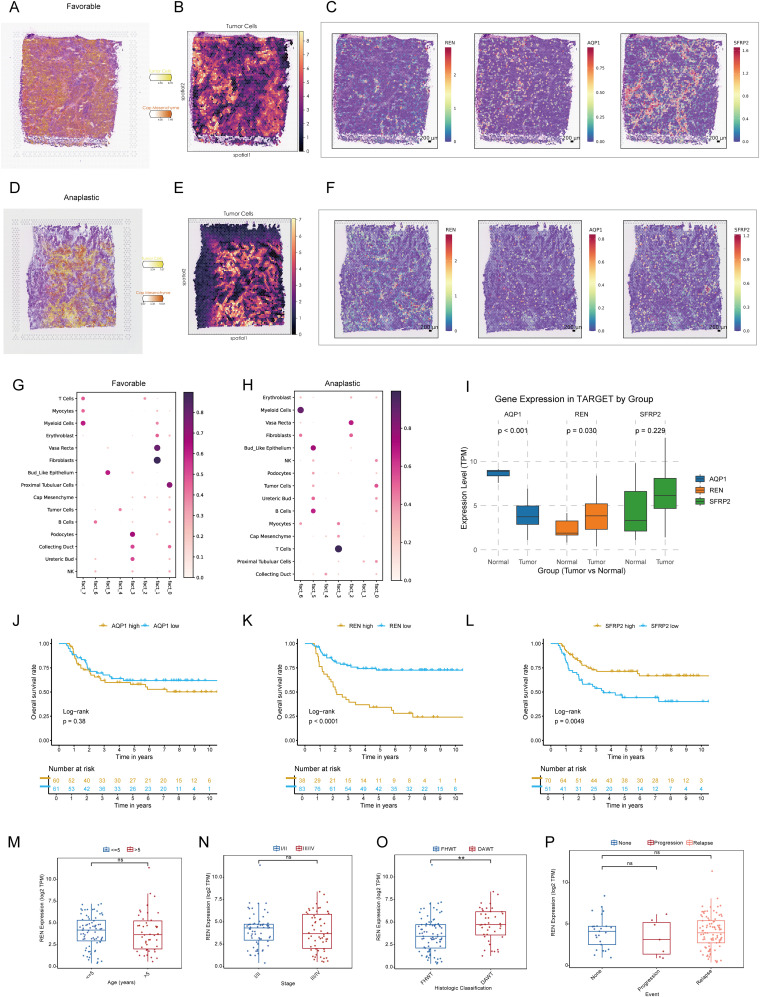
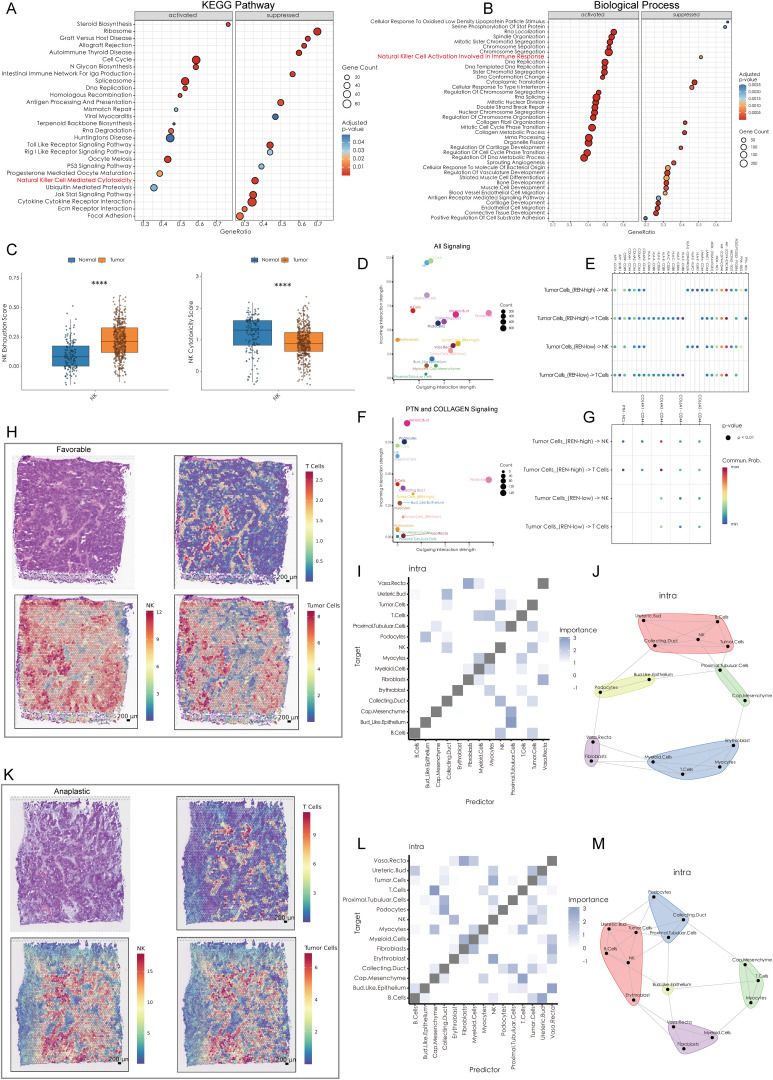
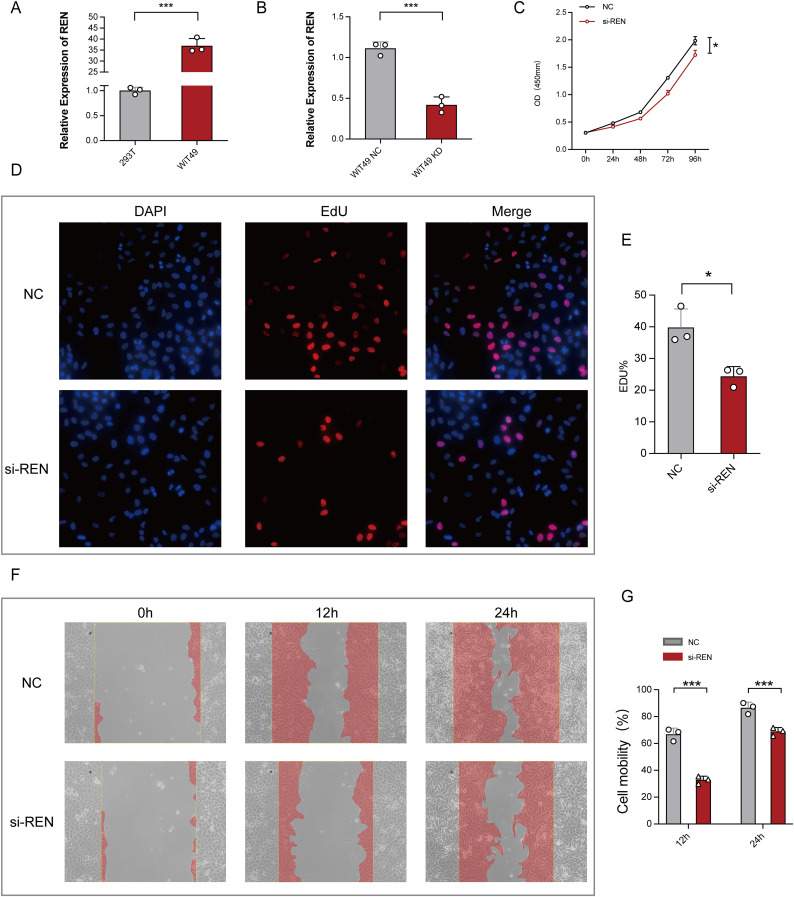
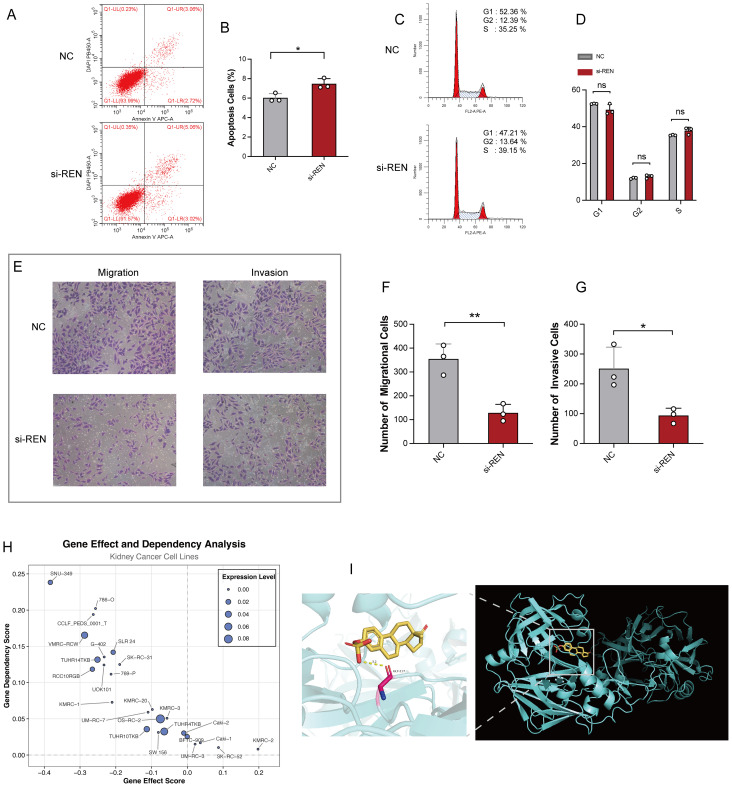
Results: Renin gene (REN) emerged as a central regulator of tumor stemness and immune evasion in WT. High-CSPI tumors exhibited enhanced tumor stemness phenotypes, metabolic reprogramming (ROS/oxidative phosphorylation), and suppressed immune activity. Spatial transcriptomics revealed distinct histological subtype-specific localization of stemness-related gene expression and physical proximity between REN-expressing tumor cells and natural killer (NK) cells. At spatial and single-cell resolution, REN-expressing tumor cells promoted NK cell exhaustion via PTN-NCL and COL4A1-CD44 ligand-receptor interactions, while showing limited impact on T cell dysfunction. Molecular docking identified estrogen-based compounds as potential REN inhibitors. Functional assays validated REN knockdown as significantly impairing tumor proliferation, migration, and survival in vitro.
Discussion: This study establishes REN as a pivotal driver of tumor stemness and immune evasion in WT, playing a dual role in promoting tumor aggressiveness and suppressing NK-mediated immune surveillance. Targeting REN offers promising therapeutic opportunities for high-risk WT cases by simultaneously inhibiting tumor progression and restoring immune function. These findings emphasize REN's potential as a transformative target for precision oncology and underscore the value of integrative transcriptomics in advancing personalized cancer treatment strategies.
Keywords: Wilms tumor; cancer stemness prognostic index; natural killer cell evasion; renin gene; tumor microenvironment; tumor stemness.
Copyright © 2025 Cao, Li, Zou, Xu, Tang and Chen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Hol JA, Jewell R, Chowdhury T, Duncan C, Nakata K, Oue T, et al. Wilms tumour surveillance in at-risk children: Literature review and recommendations from the SIOP-Europe Host Genome Working Group and SIOP Renal Tumour Study Group. Eur J Cancer. (2021) 153:51–63. doi: 10.1016/j.ejca.2021.05.014 - DOI - PubMed
-
- Perlman EJ, Grundy PE, Anderson JR, Jennings LJ, Green DM, Dome JS, et al. WT1 mutation and 11P15 loss of heterozygosity predict relapse in very low-risk wilms tumors treated with surgery alone: A children’s oncology group study. J Clin Oncol. (2011) 29:698–703. doi: 10.1200/JCO.2010.31.5192 - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous