

Mechanism of cell death and its application in the repair of inflammatory bowel disease by mesenchymal stem cells
- PMID: 40534879
- PMCID: PMC12174397
- DOI: 10.3389/fimmu.2025.1597462
Mechanism of cell death and its application in the repair of inflammatory bowel disease by mesenchymal stem cells
Abstract
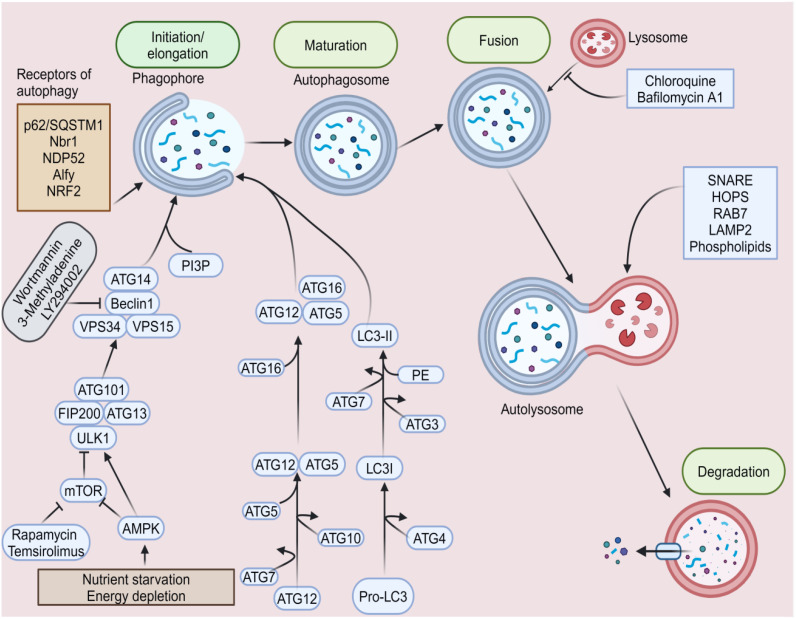
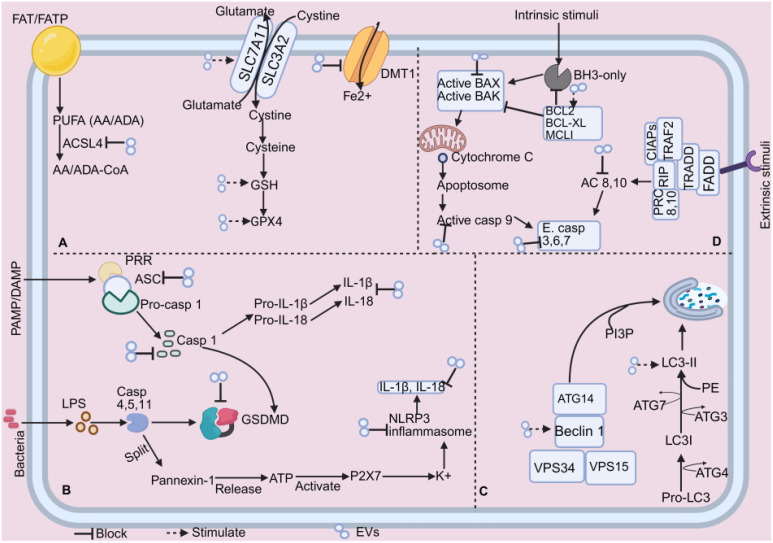
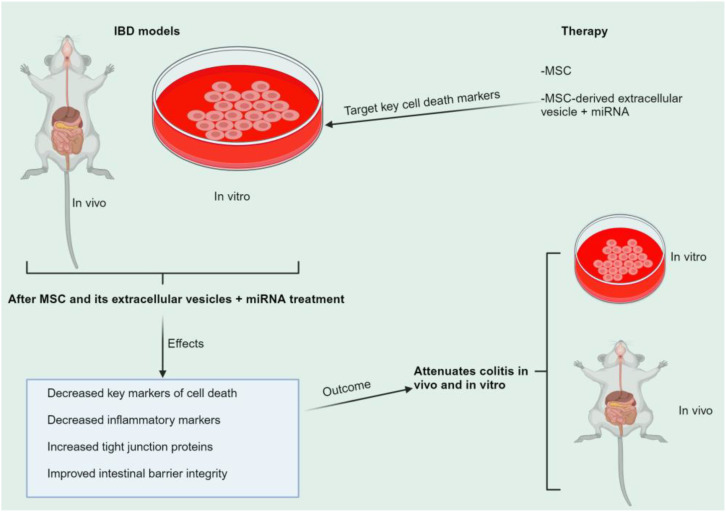
The onset and progression of inflammatory bowel disease (IBD), which encompasses ulcerative colitis and Crohn's disease, are influenced by the immune system, environmental factors, genetics, and intestinal flora. Cell death is a biological phenomenon that occurs in all living organisms; nevertheless, excessive cell death has been linked to IBD, including increased immune and intestinal epithelial cell death and intestinal barrier abnormalities. Anti-tumor necrosis factor medication, which has made significant progress in treating IBD cell death, may fail in some individuals or lose effectiveness over time, necessitating the search for a safe and effective treatment. One of the novel and emerging areas in regenerative and nanomedicine used to regulate cell death is mesenchymal stem cells (MSCs) and their mediators (extracellular vesicles). MSCs and their mediators have been found to attenuate cell death in several illnesses, including IBD. This review explores cell death mechanisms and their implications in IBD, focusing on the potential ameliorative effects of MSCs and their mediators on cell death.
Keywords: IBD; MSc; cell death; exosome; extracellular vesicles.
Copyright © 2025 Akanyibah, He, Cai, Wang, Wang and Mao.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures




References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources