

Metagenomics-assembled genomes reveal microbial metabolic adaptation to athalassohaline environment, the case Lake Barkol, China
- PMID: 40535002
- PMCID: PMC12174138
- DOI: 10.3389/fmicb.2025.1550346
Metagenomics-assembled genomes reveal microbial metabolic adaptation to athalassohaline environment, the case Lake Barkol, China
Abstract
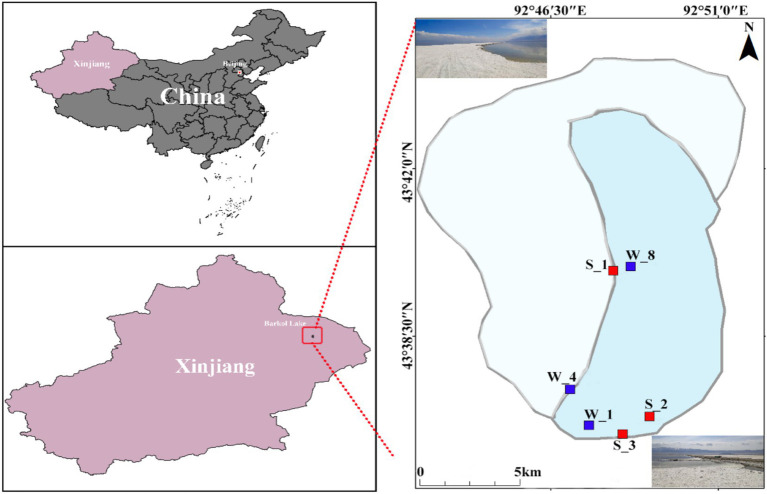
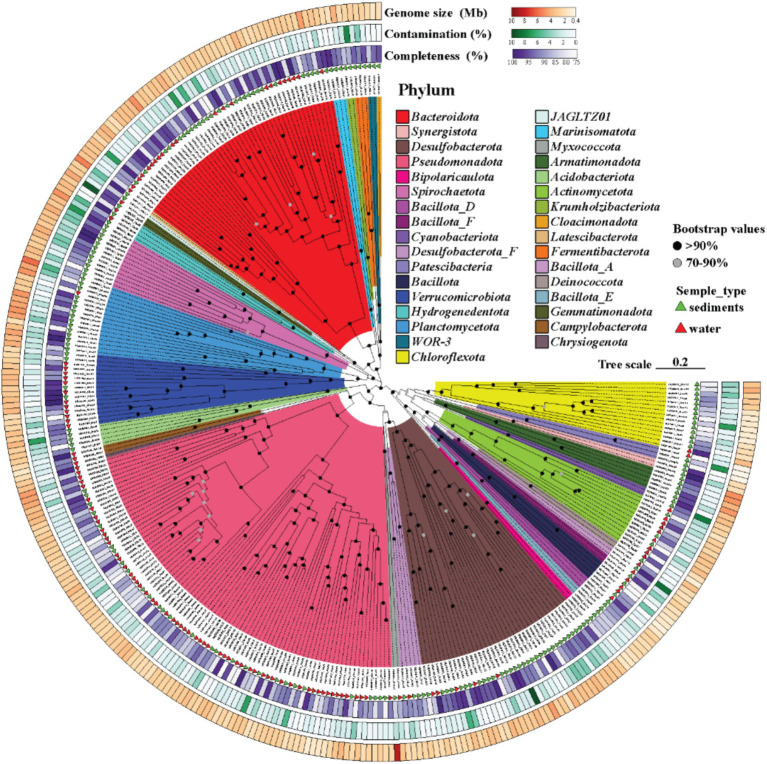
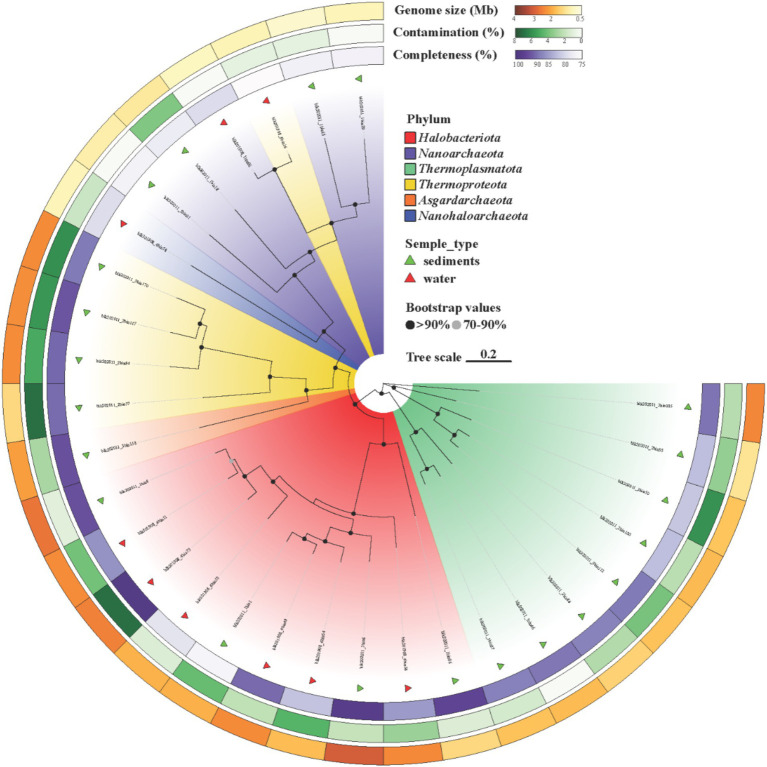
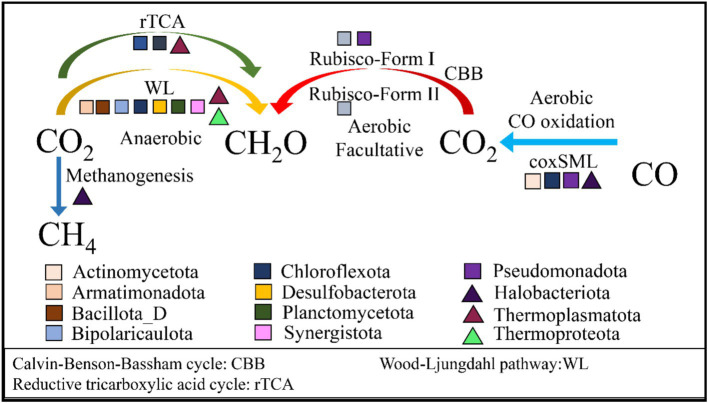
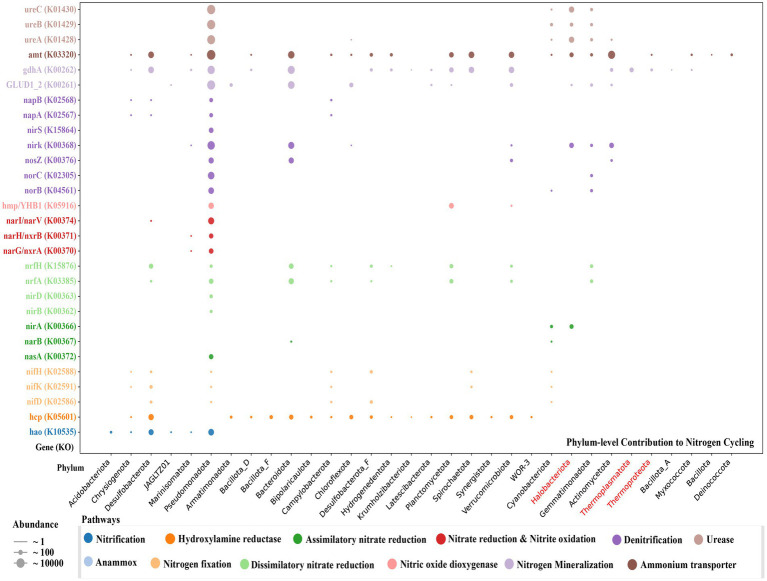
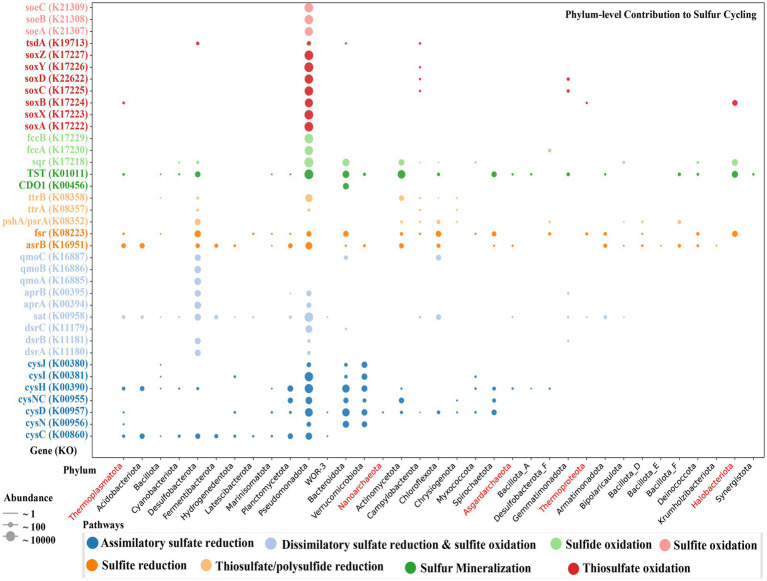
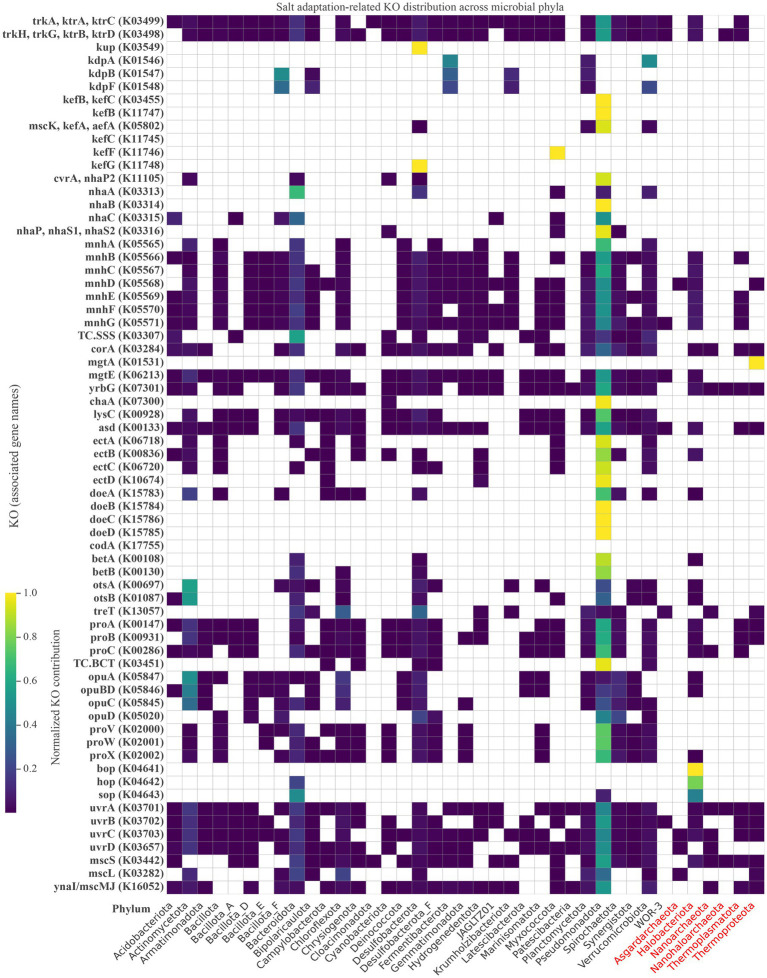
Salt-tolerant and halophilic microorganisms are critical drivers of ecosystem stability and biogeochemical cycling in athalassohaline environments. Lake Barkol, a high-altitude inland saline lake, provides a valuable natural setting for investigating microbial community dynamics and adaptation mechanisms under extreme salinity. In this study, we employed high-throughput metagenomic sequencing to characterize the taxonomic composition, metabolic potential, and ecological functions of microbial communities in both water and sediment samples from Lake Barkol. We reconstructed 309 metagenome-assembled genomes (MAGs), comprising 279 bacterial and 30 archaeal genomes. Notably, approximately 97% of the MAGs could not be classified at the species level, indicating substantial taxonomic novelty in this ecosystem. Dominant bacterial phyla included Pseudomonadota, Bacteroidota, Desulfobacterota, Planctomycetota, and Verrucomicrobiota, while archaeal communities were primarily composed of Halobacteriota, Thermoplasmatota, and Nanoarchaeota. Metabolic reconstruction revealed the presence of diverse carbon fixation pathways, including the Calvin-Benson-Bassham (CBB) cycle, the Arnon-Buchanan reductive tricarboxylic acid (rTCA) cycle, and the Wood-Ljungdahl pathway. Autotrophic sulfur-oxidizing bacteria, alongside members of Cyanobacteria and Desulfobacterota, were implicated in primary production and carbon assimilation. Nitrogen metabolism was predominantly mediated by Gammaproteobacteria, with evidence for both nitrogen fixation and denitrification processes. Sulfur cycling was largely driven by Desulfobacterota and Pseudomonadota, contributing to sulfate reduction and sulfur oxidation pathways. Microbial communities exhibited distinct osmoadaptation strategies. The "salt-in" strategy was characterized by ion transport systems such as Trk/Ktr potassium uptake and Na+/H+ antiporters, enabling active intracellular ion homeostasis. In contrast, the "salt-out" strategy involved the biosynthesis and uptake of compatible solutes including ectoine, trehalose, and glycine betaine. These strategies were differentially enriched between water and sediment habitats, suggesting spatially distinct adaptive responses to local salinity gradients and nutrient regimes. Additionally, genes encoding microbial rhodopsins were widely distributed, suggesting that rhodopsin-based phototrophy may contribute to supplemental energy acquisition under osmotic stress conditions. The integration of functional and taxonomic data highlights the metabolic versatility and ecological roles of microbial taxa in sustaining biogeochemical processes under hypersaline conditions. Overall, this study reveals extensive taxonomic novelty and functional plasticity among microbial communities in Lake Barkol and underscores the influence of salinity in structuring microbial assemblages and metabolic pathways in athalassohaline ecosystems.
Keywords: athalassohaline Lake Barkol; metabolic adaptation; metagenome-assembled genomes; microbial consortia; osmoadaptation strategies.
Copyright © 2025 Xamxidin, Zhang, Zheng, Chen and Wu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
-
- Alamoudi R., Barozzi A., Michoud G., van Goethem M. W., Odobel C., Chen Y., et al. (2025). Metabolic redundancy and specialisation of novel sulfide-oxidizing Sulfurimonas and Sulfurovum along the brine-seawater interface of the Kebrit deep. Environ. Microbiome 20:19. doi: 10.1186/s40793-025-00669-7, PMID: - DOI - PMC - PubMed
-
- Andrews S. FastQC: A quality control tool for high throughput sequence data. (2010). Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
LinkOut - more resources
Full Text Sources