

The innate immune receptor NLRX1 is a novel required modulator for mPTP opening: implications for cardioprotection
- PMID: 40536683
- PMCID: PMC12325489
- DOI: 10.1007/s00395-025-01124-x
The innate immune receptor NLRX1 is a novel required modulator for mPTP opening: implications for cardioprotection
Abstract
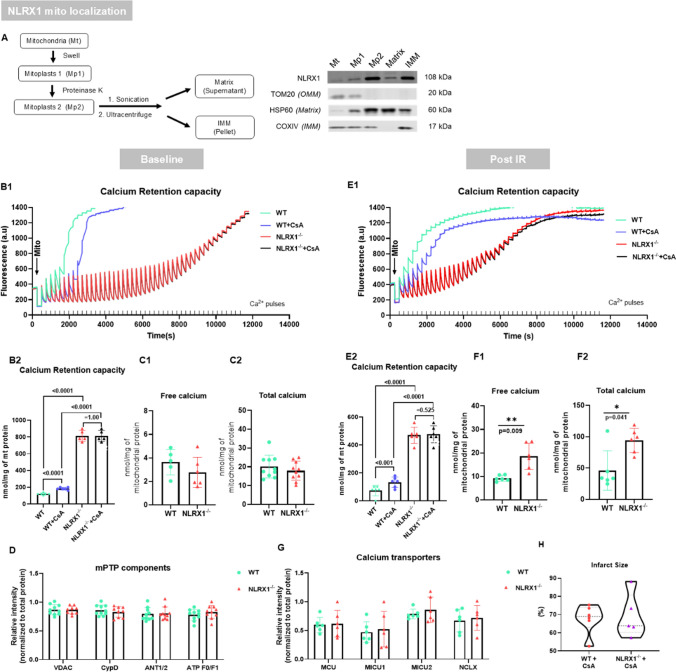
NLRX1 is the only NOD-like innate immune receptor that localises to mitochondria. We previously demonstrated that NLRX1 deletion increased infarct size in isolated mouse hearts subjected to ischemia-reperfusion injury (IRI); however, underlying mechanisms are yet to be identified. Given the crucial role played by mitochondria in cardiac IRI, we here hypothesise that NLRX1 affects key mechanisms of cardiac IRI. Cardiac IRI was evaluated in isolated C57BL/6J (WT) and NLRX1 knock out (KO) mouse hearts. The following known modulators of IRI were explored in isolated hearts, isolated mitochondria; or permeabilised cardiac fibres: 1) mTOR/RISK/autophagy regulation, 2) AMPK and mitochondrial energy production, and 3) mitochondrial permeability transition pore (mPTP) opening. NLRX1 deletion increased IRI, and cardiac NLRX1 was decreased after IRI in mouse and pig hearts. NLRX1 ablation caused decreased mTOR and RISK pathway (Akt, ERK, and S6K) activation following IR, without affecting autophagy/inflammation/oxidative stress markers. The RISK activator Urocortin dissipated NLRX1 effects on mTOR, RISK pathway and IRI, indicating that increased cardiac IRI with NLRX1 deletion is, at least partly, due to impaired RISK activation. The energy sensor AMPK was activated in NLRX1 KO hearts, possibly due to slowed mitochondrial respiratory responses (impaired mitochondrial permeability) towards palmitoylcarnitine in permeabilised cardiac fibres. NLRX1 deletion completely abolished calcium-induced mPTP opening, and cyclosporine A (CsA) effects on mPTP, both before and after IR, and was associated with increased mitochondrial calcium content after IR. Mitochondrial sub-fractionation studies localised NLRX1 to the inner mitochondrial membrane. NLRX1 deletion associated with decreased phosphorylation of mitochondrial Got2, Cx43, Myl2, Ndufb7 and MICOS10. The mPTP inhibitor CsA abolished IRI differences between KO and WT hearts, suggesting that the permanent closure of mPTP due to NLRX1 deletion contributed to the increased IR sensitivity of NLRX1 KO hearts. This is the first demonstration that the mitochondrial NLRX1 is a novel factor required for mPTP opening and contributes to cardioprotection against acute IRI through RISK pathway activation and prevention of permanent mPTP closure.
Keywords: AMPK; I/R injury; Mitochondria; Mitochondrial transition pore opening; NLRX1; RISK pathway.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Conflict of interest: PF is the founder and CEO of Pharmahungary Group, a group of R&D companies www.pharmahungary.com . All other authors report no conflict of interest.
Figures





References
-
- Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA Jr, Jonas EA (2014) An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A 111:10580–10585. 10.1073/pnas.1401591111 - PMC - PubMed
-
- Altschuld RA, Hohl CM, Castillo LC, Garleb AA, Starling RC, Brierley GP (1992) Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am J Physiol 262:H1699-1704. 10.1152/ajpheart.1992.262.6.H1699 - PubMed
-
- Arslan F, Houtgraaf JH, Keogh B, Kazemi K, de Jong R, McCormack WJ, O’Neill LA, McGuirk P, Timmers L, Smeets MB, Akeroyd L, Reilly M, Pasterkamp G, de Kleijn DP (2012) Treatment with OPN-305, a humanized anti-Toll-Like receptor-2 antibody, reduces myocardial ischemia/reperfusion injury in pigs. Circ Cardiovasc Interv 5:279–287. 10.1161/CIRCINTERVENTIONS.111.967596 - PubMed
-
- Arslan F, Smeets MB, O’Neill LA, Keogh B, McGuirk P, Timmers L, Tersteeg C, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP (2010) Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation 121:80–90. 10.1161/CIRCULATIONAHA.109.880187 - PubMed
-
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434:658–662. 10.1038/nature03434 - PubMed
MeSH terms
Substances
Grants and funding
- 91818602/NWO
- 91818602/NWO-ZonMW
- 2021/Dutch Cardiovascular Alliance
- 20CVD01/Fondation Leducq
- 16cvd04/Fondation Leducq
- 739593/HORIZON EUROPE European Innovation Council
- unkp-22-4-ii-se-3/New National ExcellenceProgrm of the Ministry of Human Capacities
- 2021/38315/ZONMW/ZON-MW
- EU-Cardioprotection CA16225/Horizon 2020 Framework Programme
- EU-Cardioprotection CA16225/HORIZON EUROPE Framework Programme
- 2018/EFSD
- research grant/Boehringer Ingelheim Stiftung
- 82400401/National Natural Science Foundation of China
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
