

Retinoma: An overview
- PMID: 40539010
- PMCID: PMC12175641
- DOI: 10.1002/ped4.12472
Retinoma: An overview
Abstract
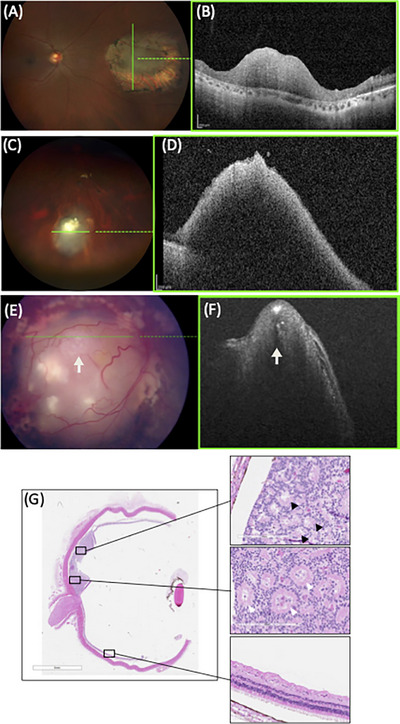
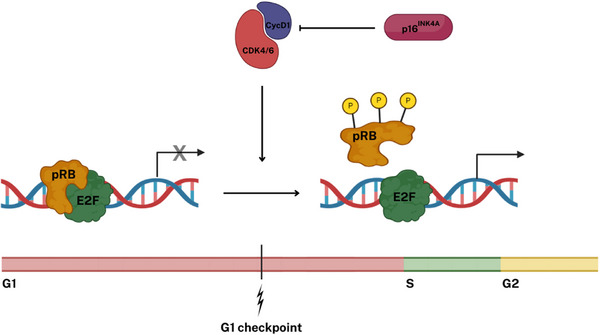
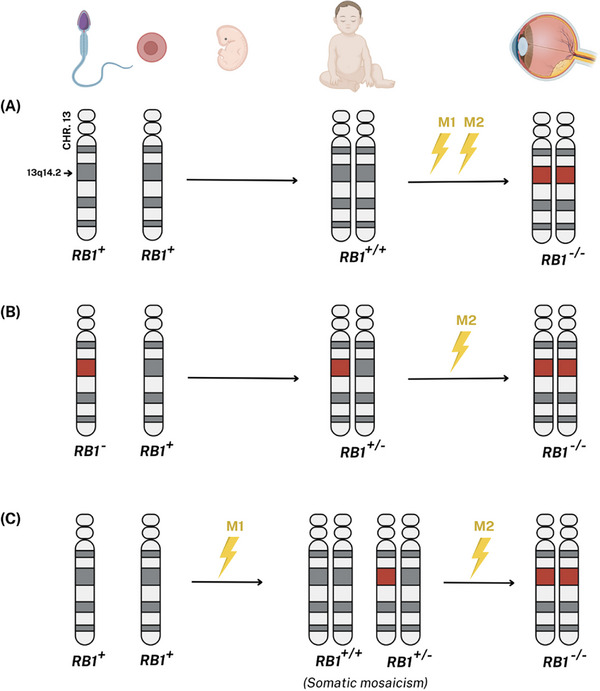
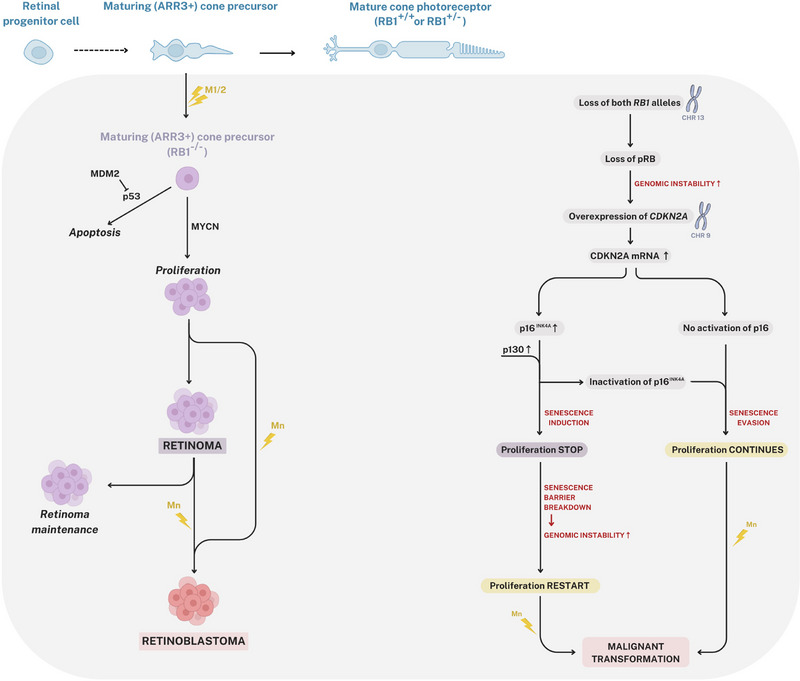
Retinoma, also referred to as retinocytoma, is a benign manifestation of biallelic retinoblastoma gene (RB1) inactivation. Genetic or epigenetic loss of retinoblastoma protein in maturing cone precursors induces genomic instability which leads to upregulation of senescence-associated p16INK4a and p130, resulting in non-proliferative retinoma. When senescence pathways fail and genetic instability accumulates to a critical level through altered gene copies of oncogenes and tumor suppression genes, transformation into RB1 -/- retinoblastoma occurs. Thus, the management of retinoma involves frequent ophthalmic examination and imaging to monitor the size and characteristics of the tumor, ensure stability, and rule out malignant transformation. Key ophthalmoscopic features of retinoma often include a translucent whitish-gray retinal mass, calcification, retinal pigment epithelial alterations with well-defined margins, located typically around the lesion, as well as a zone of chorioretinal atrophy. This review aims to provide a comprehensive overview of this non-malignant tumor drawing from current understanding of its molecular genetics, clinical characteristics, diagnostic modalities, differential diagnosis, management, and prognosis. A deeper understanding of retinoma could offer valuable insights into how retinoblastoma develops and oncogenesis more broadly, paving the way for improved strategies to prevent and treat this malignant tumor.
Keywords: Ocular oncology; Retinal tumor; Retinoblastoma; Retinocytoma; Retinoma.
© 2025 The Author(s). Pediatric Investigation published by John Wiley & Sons Australia, Ltd on behalf of Futang Research Center of Pediatric Development.
Conflict of interest statement
None.
Figures
Similar articles
-
Comprehensive mutational profiling identifies new driver events in cutaneous leiomyosarcoma.Br J Dermatol. 2025 Jan 24;192(2):335-343. doi: 10.1093/bjd/ljae386. Br J Dermatol. 2025. PMID: 39392932
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
Survivor, family and professional experiences of psychosocial interventions for sexual abuse and violence: a qualitative evidence synthesis.Cochrane Database Syst Rev. 2022 Oct 4;10(10):CD013648. doi: 10.1002/14651858.CD013648.pub2. Cochrane Database Syst Rev. 2022. PMID: 36194890 Free PMC article.
-
Molecular feature-based classification of retroperitoneal liposarcoma: a prospective cohort study.Elife. 2025 May 23;14:RP100887. doi: 10.7554/eLife.100887. Elife. 2025. PMID: 40407808 Free PMC article.
-
Cost-effectiveness of using prognostic information to select women with breast cancer for adjuvant systemic therapy.Health Technol Assess. 2006 Sep;10(34):iii-iv, ix-xi, 1-204. doi: 10.3310/hta10340. Health Technol Assess. 2006. PMID: 16959170
References
-
- Gallie BL, Dupont B, Whitsett C, Kitchen FD, Ellsworth RM, Good RA. Histocompatibility typing in spontaneous regression of retinoblastoma. Prog Clin Biol Res. 1977;16:229‐237. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous