

Comparative analysis of codon usage bias and host adaptation across avian metapneumovirus genotypes
- PMID: 40541102
- PMCID: PMC12214266
- DOI: 10.1016/j.psj.2025.105428
Comparative analysis of codon usage bias and host adaptation across avian metapneumovirus genotypes
Abstract
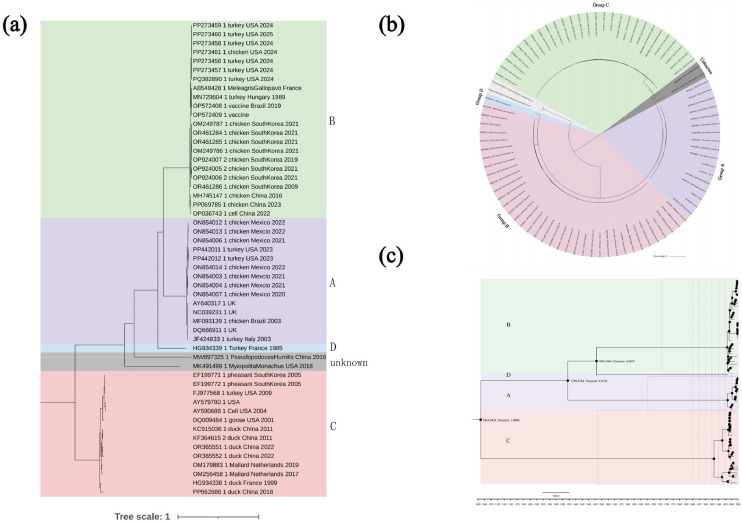
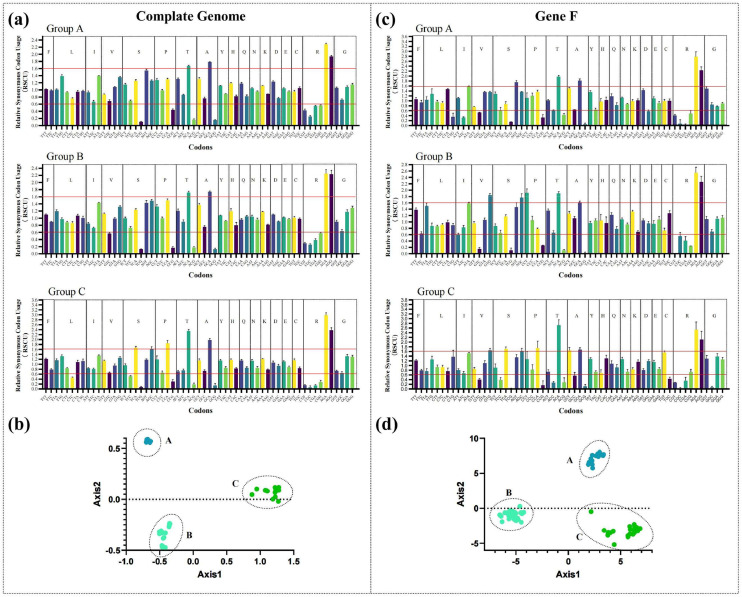
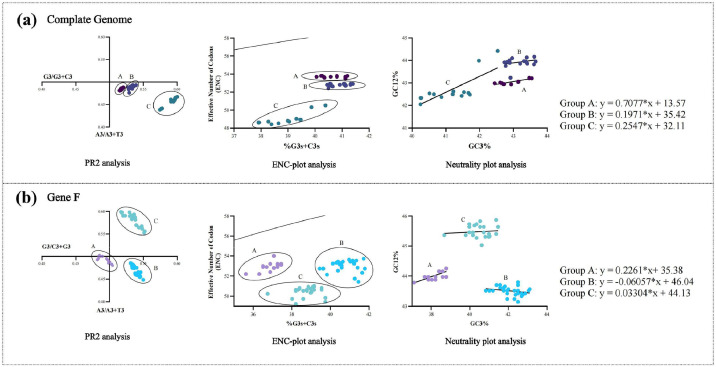
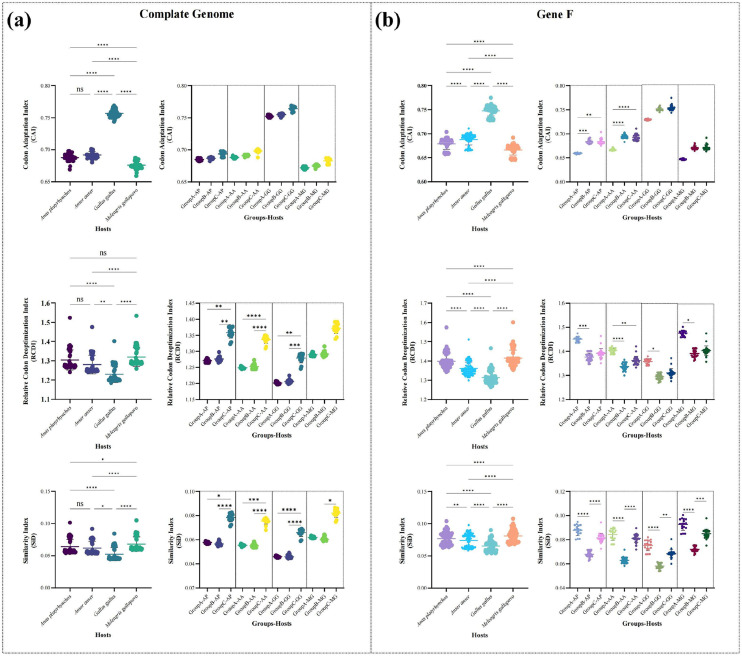
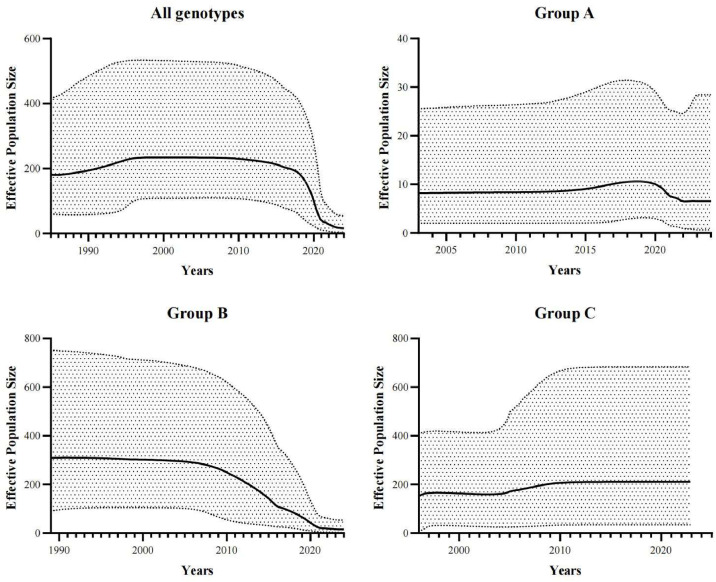
Avian metapneumovirus (aMPV) is a significant pathogen affecting poultry worldwide, causing respiratory disease and economic losses. This study investigated the genetic and evolutionary differences among aMPV genotypes through codon usage bias analysis. Using whole-genome and F gene sequences, we assessed phylogenetic relationships, codon usage patterns, evolutionary pressures, and host adaptation. Our results indicate clear genotype differentiation in the phylogenetic tree, with Group C identified as the earliest diverging lineage of aMPV. The F gene exhibits independent evolutionary trajectories, reflecting distinct selective pressures. Codon usage bias varies across genotypes and is primarily driven by selection pressure, with Groups B and C experiencing stronger selective constraints. The F gene, crucial for viral entry and adaptation, undergoes intense selection, optimising codon usage for host adaptation. Host adaptation analysis reveals that aMPV is most suited to chickens. Additionally, Group B exhibits the largest population size; however, recent declines, particularly in this genotype, suggest that vaccine-driven selection pressure may be influencing aMPV population dynamics. These findings provide critical insights into aMPV evolution, highlighting the role of codon usage bias and selection pressure in shaping viral adaptation. Understanding these evolutionary mechanisms may aid in vaccine development and disease control strategies.
Keywords: Avian metapneumovirus; Codon usage bias; Genotype; Host adaptation; Phylogenetic analysis.
Copyright © 2025. Published by Elsevier Inc.
Conflict of interest statement
Declaration of competing interest The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Shuiqin Shi reports financial support was provided by Anqing Normal University. If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures

References
-
- Bao Y., Yu M., Liu P., Hou F., Muhammad F., Wang Z., Li X., Zhang Z., Wang S., Chen Y., Cui H., Liu A., Qi X., Pan Q., Zhang Y., Gao L., Li K., Liu C., He X., Wang X., Gao Y. Novel inactivated subtype B avian metapneumovirus vaccine induced humoral and cellular immune responses. Vaccines (Basel) 2020;8 doi: 10.3390/vaccines8040762. - DOI - PMC - PubMed
-
- Brown P.A., Lemaitre E., Briand F.X., Courtillon C., Guionie O., Allee C., Toquin D., Bayon-Auboyer M.H., Jestin V., Eterradossi N. Molecular comparisons of full length metapneumovirus (MPV) genomes, including newly determined French AMPV-C and -D isolates, further supports possible subclassification within the MPV Genus. PLoS One. 2014;9 doi: 10.1371/journal.pone.0102740. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
