

Immune targeting of triple-negative breast cancer through a clinically actionable STING agonist-CAR T cell platform
- PMID: 40543510
- PMCID: PMC12281382
- DOI: 10.1016/j.xcrm.2025.102198
Immune targeting of triple-negative breast cancer through a clinically actionable STING agonist-CAR T cell platform
Abstract
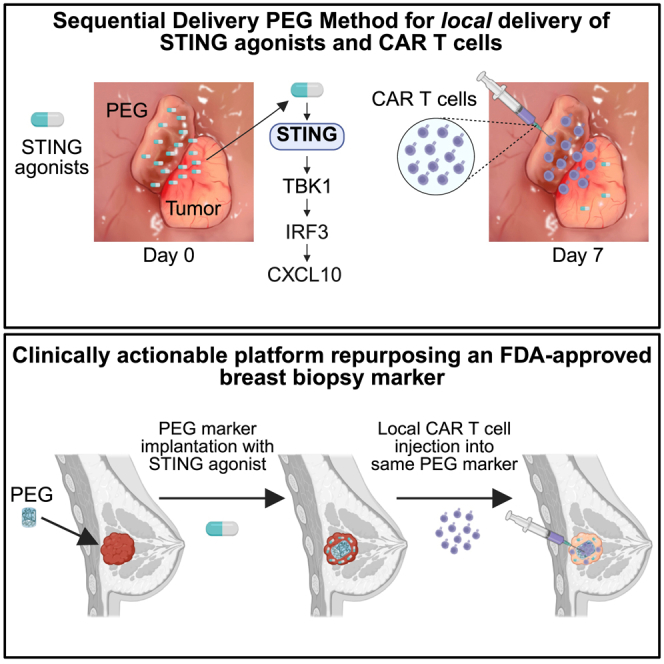
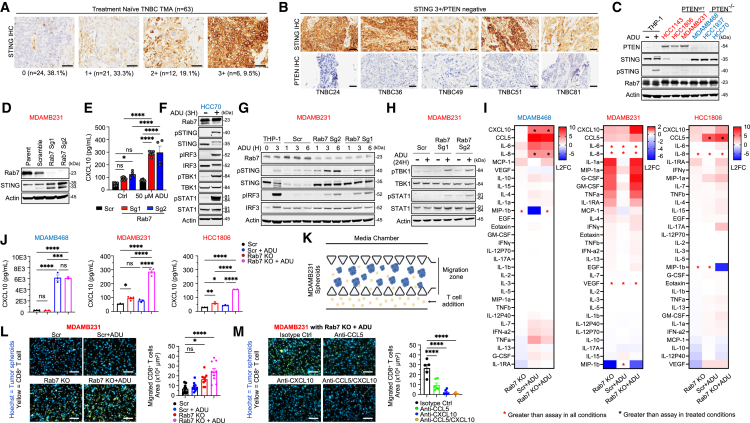
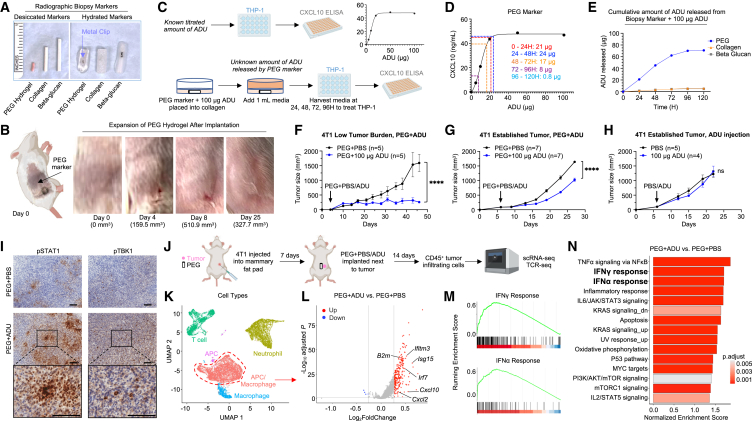
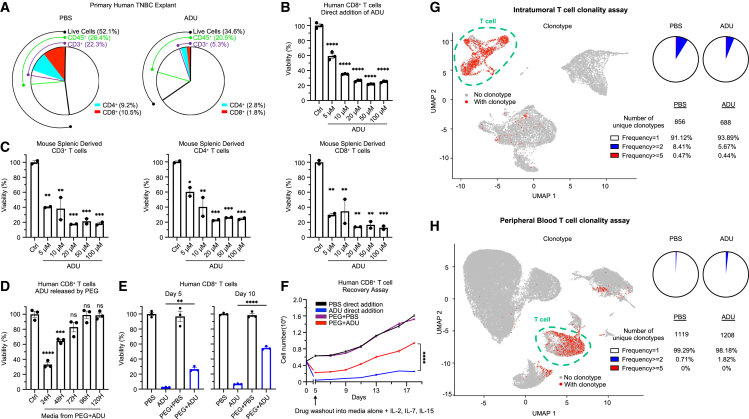
Stimulator of interferon genes (STING) has emerged as a critical cancer immunotherapy target. However, no STING agonist has advanced beyond phase I/II clinical trials, as obstacles center around applying STING agonism to the appropriate clinical context, retaining it in the tumor microenvironment (TME), and limiting its T cell toxicity. Using triple-negative breast cancer (TNBC), we identify defective STING turnover as a cancer state promoting hypersensitivity to STING agonism. We also repurpose a US Food and Drug Administration (FDA)-approved polyethylene glycol (PEG) biopsy marker to deliver STING agonists in a controlled release fashion into the TME. However, STING agonist-induced T cell toxicity limits robust endogenous clonal T cell response, which can be overcome by sequential co-delivery of the STING agonists with CAR T cell therapy using the same PEG marker, eradicating orthotopic TNBC in mouse models while also controlling distant disease. These findings identify a highly translatable platform to combine STING agonists with CAR T cell therapy locally for TNBC and potentially other solid cancers.
Keywords: CAR t cells; PEG marker; STING agonist; triple-negative breast cancer.
Copyright © 2025 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests DFCI/BWH have filed patents related to this work (inventors: T.U.B. and D.A.B.). T.U.B. consults for QIAGEN and has ownership interest in patents using STING levels as a biomarker for cancer immunotherapy and microfluidic cell culture of patient-derived tumor cell spheroids. D.A.B. is a consultant/advisory board member at QIAGEN and NMS and has ownership interest in STING biomarker and microfluidic culture patents and in Xsphera Biosciences.
Figures
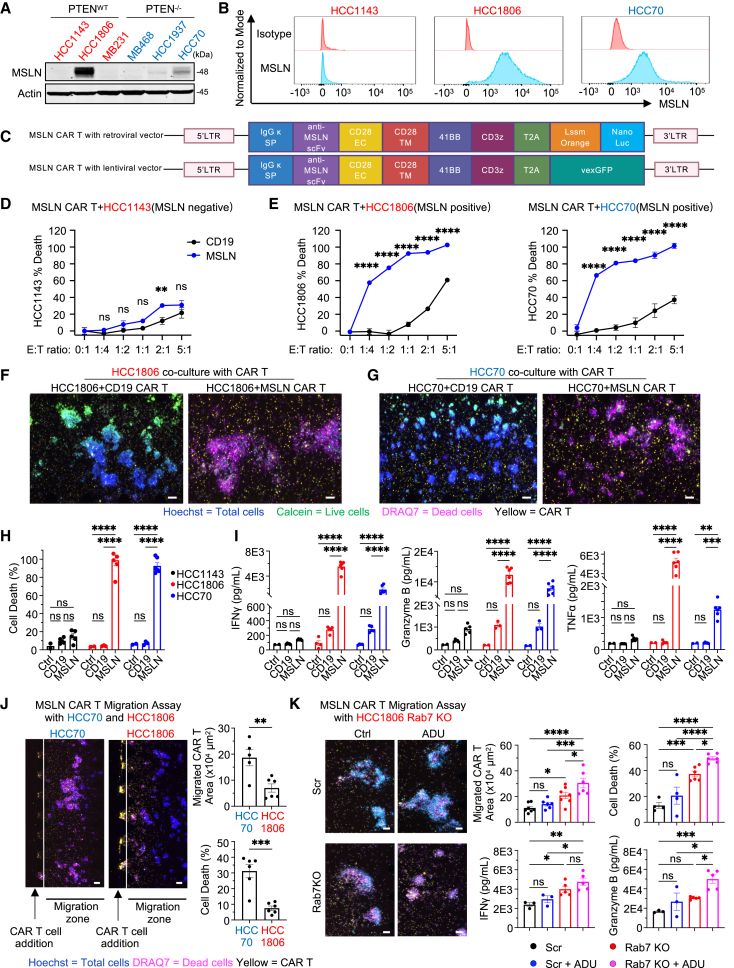
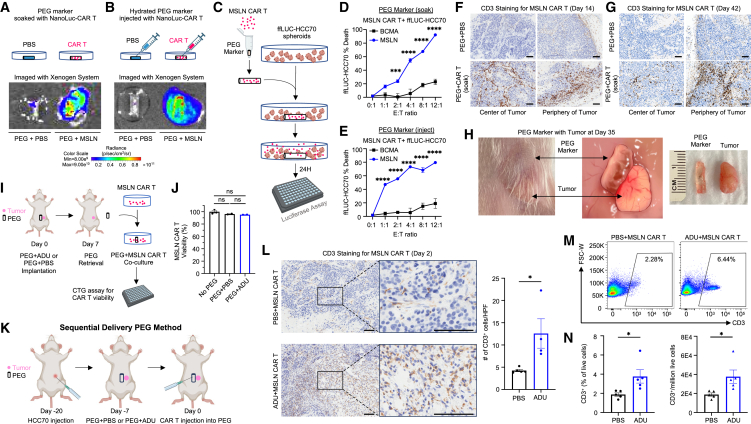
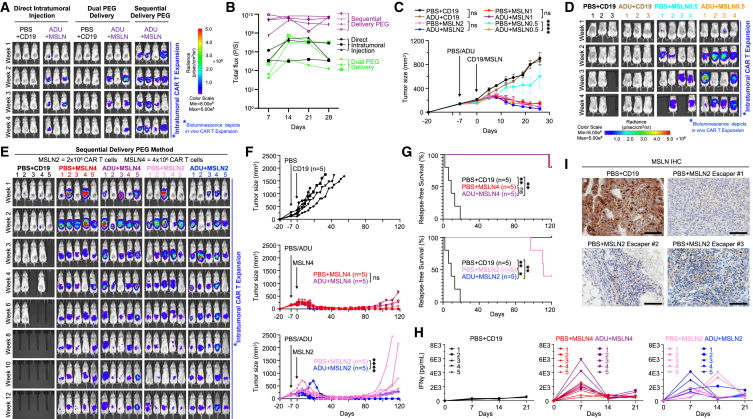
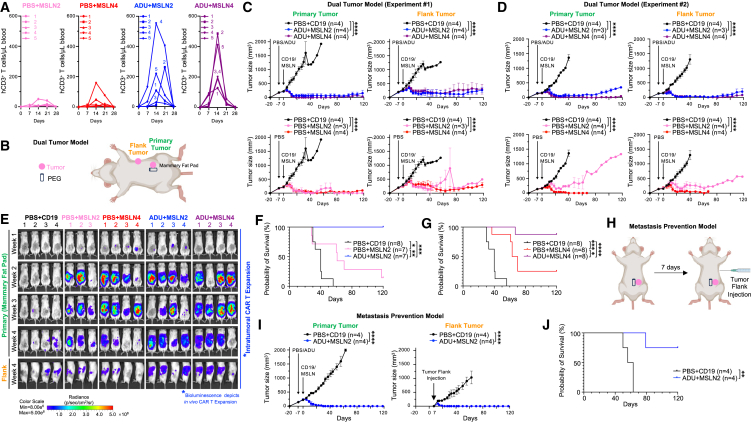
References
-
- Burstein M.D., Tsimelzon A., Poage G.M., Covington K.R., Contreras A., Fuqua S.A.W., Savage M.I., Osborne C.K., Hilsenbeck S.G., Chang J.C., et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015;21:1688–1698. doi: 10.1158/1078-0432.CCR-14-0432. - DOI - PMC - PubMed
-
- Cortes J., Cescon D.W., Rugo H.S., Nowecki Z., Im S.A., Yusof M.M., Gallardo C., Lipatov O., Barrios C.H., Holgado E., et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. 2020;396:1817–1828. doi: 10.1016/S0140-6736(20)32531-9. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
