

Development of translational read-through-inducing drugs as novel therapeutic options for patients with Fanconi anemia
- PMID: 40544182
- PMCID: PMC12182573
- DOI: 10.1038/s41420-025-02571-0
Development of translational read-through-inducing drugs as novel therapeutic options for patients with Fanconi anemia
Abstract
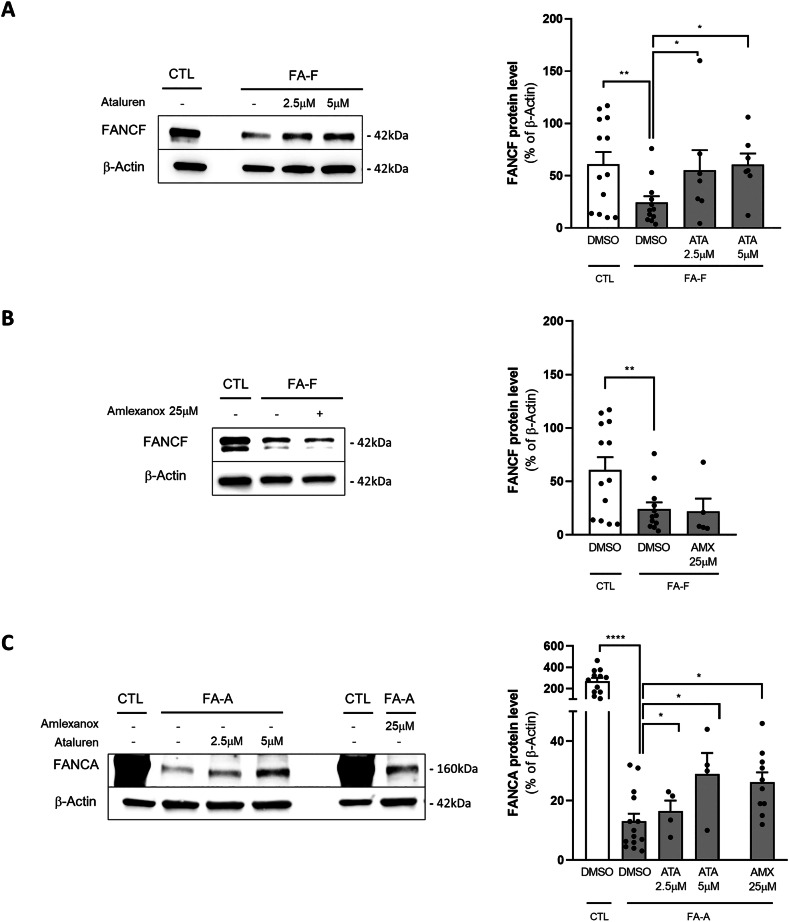
Fanconi anemia (FA) is caused by mutations affecting FANC genes involved in DNA repair, with nearly 20% of FA patients harboring nonsense mutations. Ataluren (PTC124) is a translational read-through-inducing drug (TRID) already approved in Europe that has a well-established safety profile even in pediatric patients. Amlexanox, an anti-inflammatory drug, also promotes read-through of premature stop codons caused by nonsense mutations. We compared ataluren and amlexanox in rescuing FANCA, FANCC and FANCF protein synthesis in lymphoblastoid cell lines and fibroblasts obtained from FA patients with nonsense mutations. While ataluren restored all FANC protein levels, amlexanox was partially effective only on FANCA. Notably, the rescue of FANC proteins resulted in a significant downregulation of p53. Moreover, unlike amlexanox, ataluren remarkably improved cell viability and reduced chromosomal aberrations upon exposure to genotoxic compounds. Amlexanox primarily reduced the signal transducer and activator of transcription 2 (STAT2) phosphorylation. Furthermore, FANCA-mutated fibroblasts exhibited a higher frequency of micronuclei formation as well as lower lamin B1 expression compared to their gene-edited counterpart re-expressing wild-type FANCA. Interestingly, ataluren significantly limited the generation of micronuclei in nonsense-mutated primary FANCC fibroblasts, restoring lamin B1 expression. This study represents a milestone of drug development for FA as it paves the way for clinical development of TRIDs, indicating ataluren as a promising approach to address the genetic instability and reduce the risk of malignant transformation in FA cells. Moreover, these results highlight the importance of a reliable experimental pipeline to assess whether minimal protein rescue via translational read-through can yield meaningful phenotypic rescue.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethics: Ethics approval was not needed, as this study does not involve patients or animals.
Figures







References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
