

Targeting the RAGE-RIPK1 binding site attenuates diabetes-associated cognitive deficits
- PMID: 40544260
- PMCID: PMC12182694
- DOI: 10.1186/s12974-025-03489-1
Targeting the RAGE-RIPK1 binding site attenuates diabetes-associated cognitive deficits
Abstract
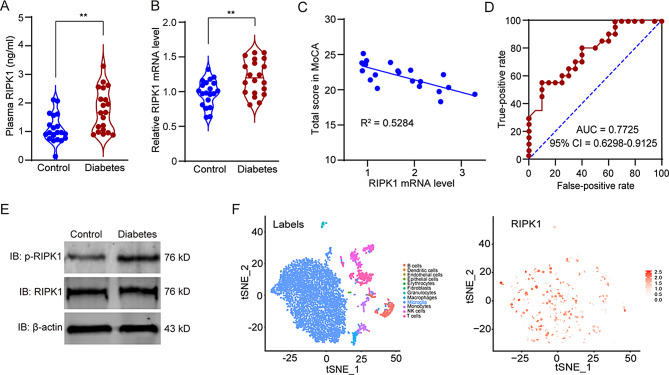
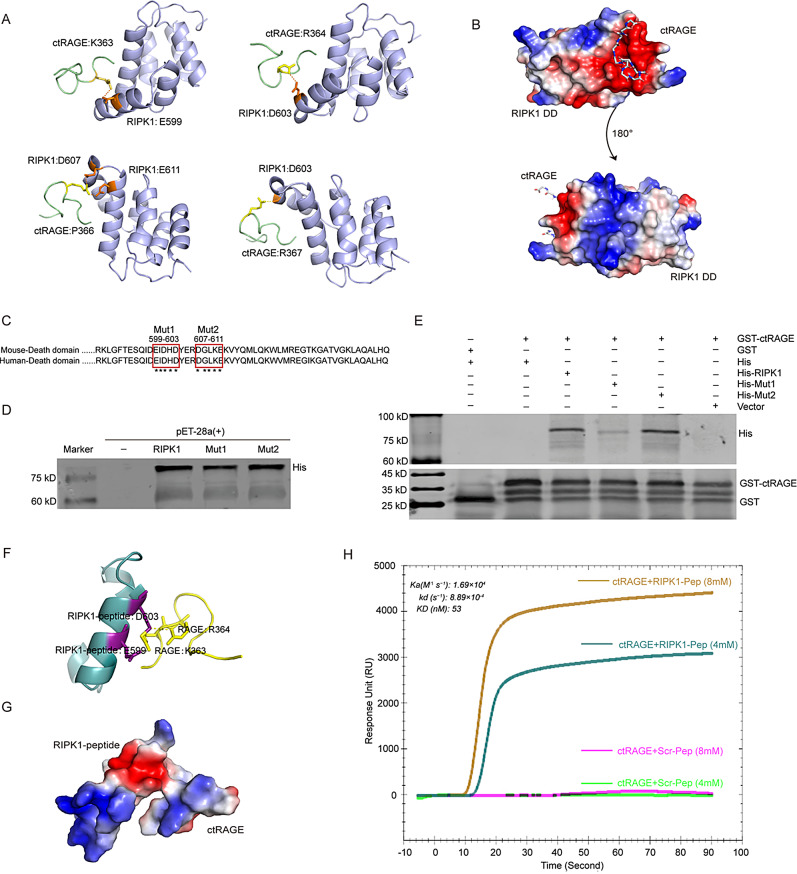
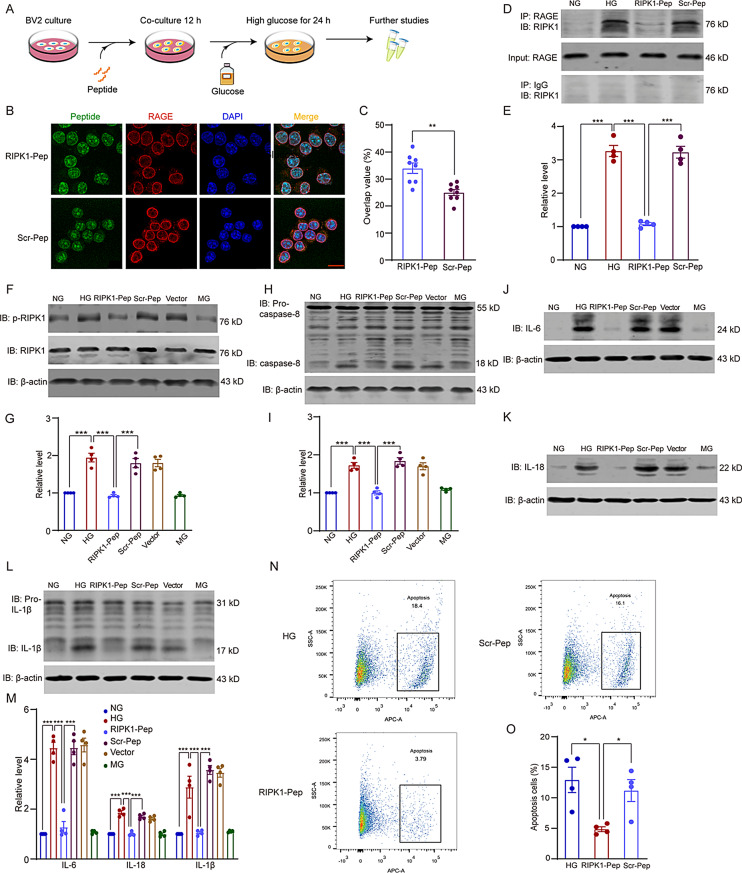
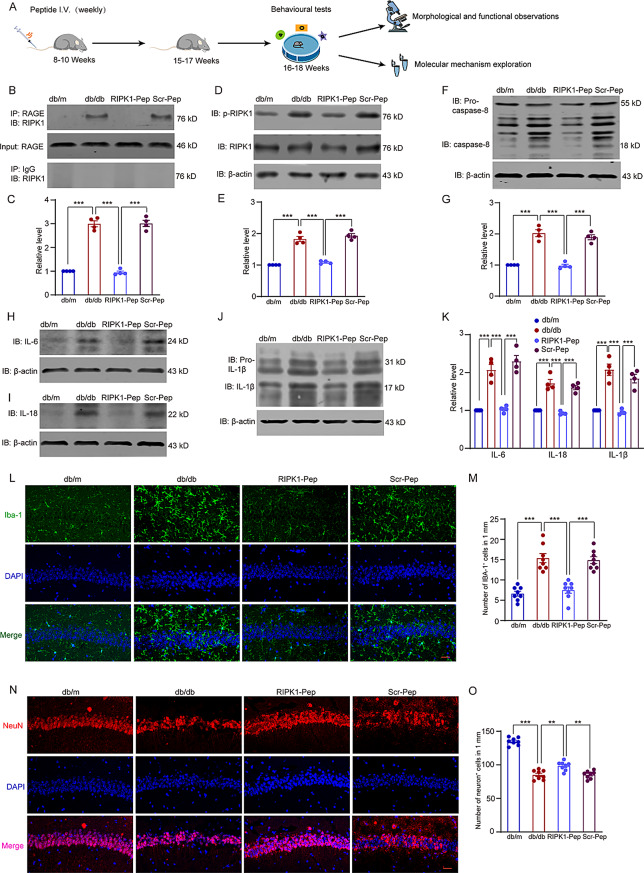
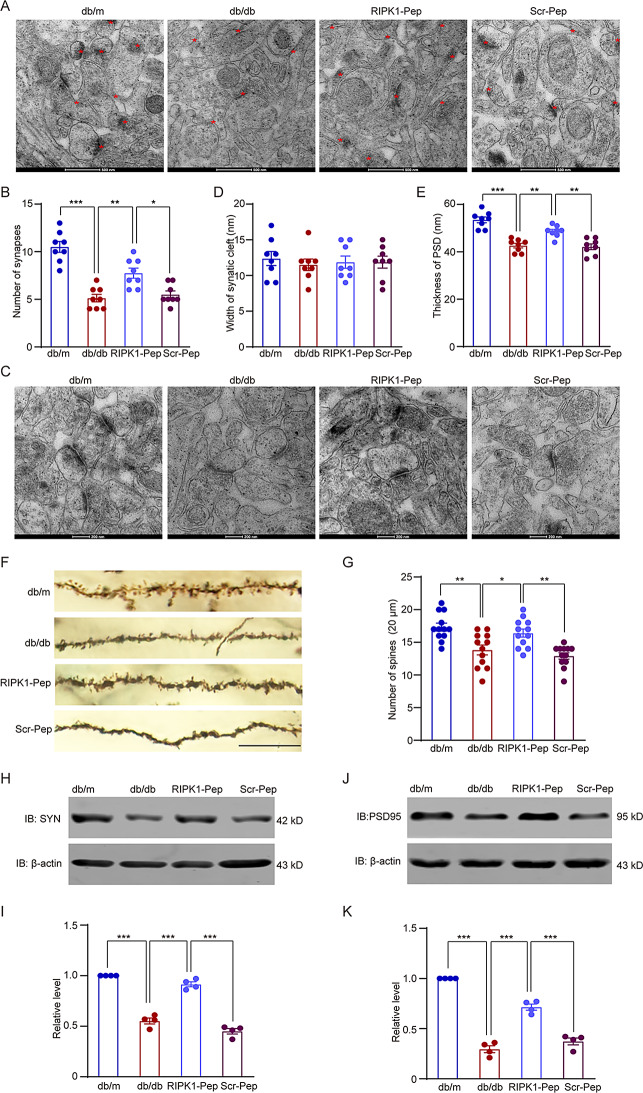
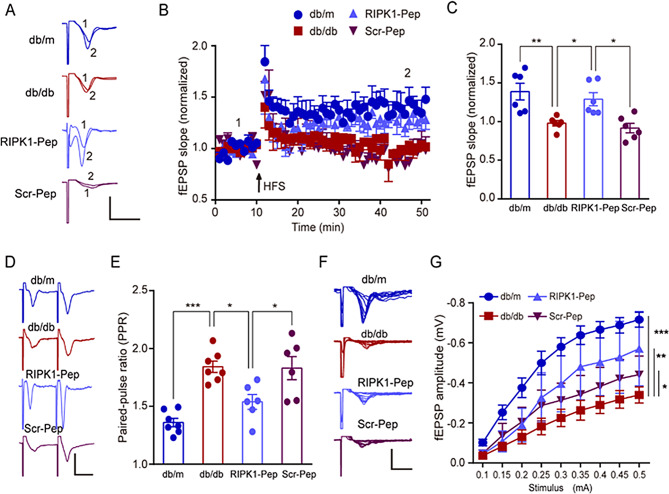
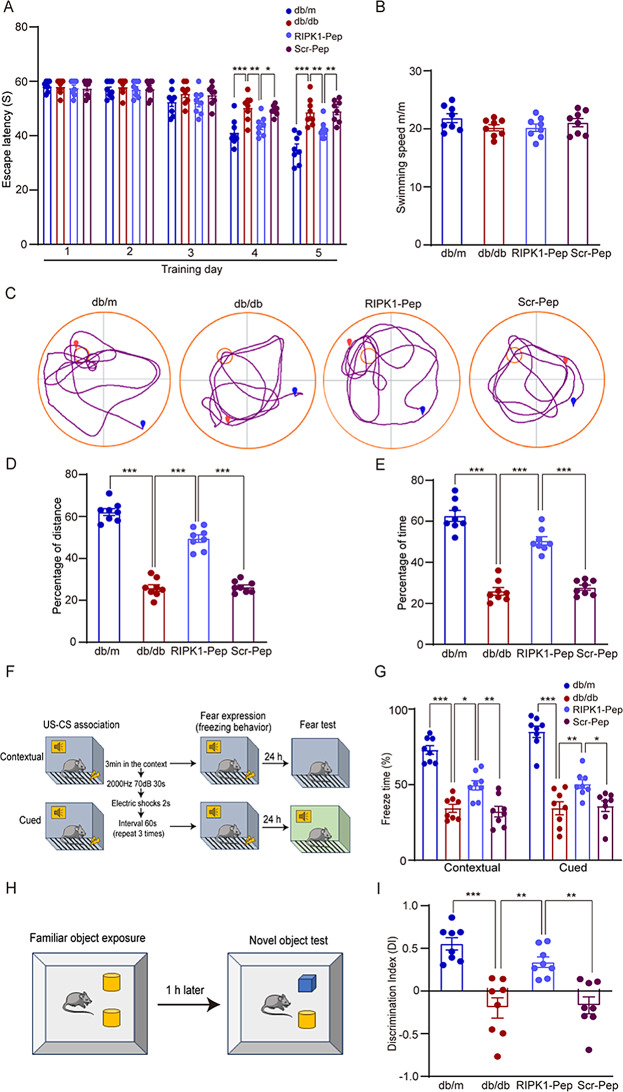
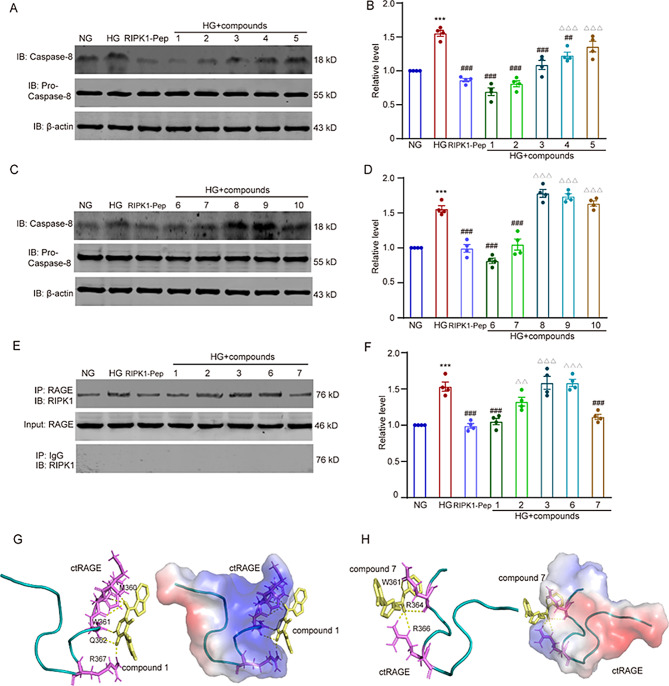
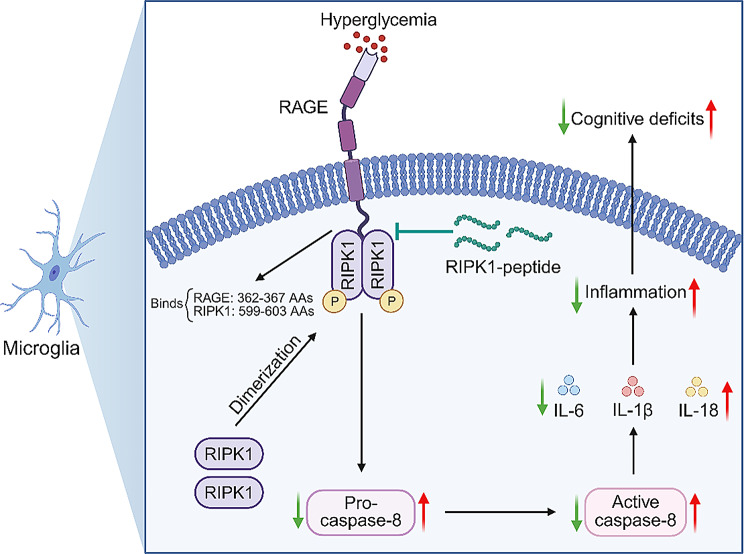
Microglial activation can cause neuroinflammation and the consequent neurological impairments play prominent roles in diabetes-associated cognitive deficits. Receptor-interacting protein kinase 1 (RIPK1) phosphorylation is involved in this deleterious microglial activation, but the exact molecular mechanisms are not clear. Here, RIPK1 expression was increased in diabetic patients with cognitive impairment. Furthermore, in diabetic mice, RIPK1 death domain directly binds to C-terminal of the receptor for advanced glycation end products (ctRAGE) could regulate RIPK1 phosphorylation in microglia. This RAGE-RIPK1 complex activates inflammatory signaling, resulting in cascades that ultimately promote cognitive impairment in diabetic mice. An engineered brain-targeting RIPK1 peptide blocked binding of RIPK1 to RAGE, which inhibited RIPK1 phosphorylation, decreased neuroinflammation, improved neuronal morphology and function, and prevented diabetes-associated cognitive deficits in mice. This study uncovers a previously unknown mechanism of neuroinflammation and suggests a novel therapeutic avenue for treating cognitive deficits induced by hyperglycemia.
Keywords: Diabetes-associated cognitive deficits; Neuroinflammation; RIPK1 peptide; Receptor-interacting protein kinase 1; The receptor for advanced glycation end products.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Binding of RAGE and RIPK1 induces cognitive deficits in chronic hyperglycemia-derived neuroinflammation.CNS Neurosci Ther. 2024 Mar;30(3):e14449. doi: 10.1111/cns.14449. Epub 2023 Sep 4. CNS Neurosci Ther. 2024. PMID: 37665158 Free PMC article.
-
miR-214 and Its Primary Transcript Dnm3os Regulate Fibrosis and Inflammation Through RAGE Signaling in Diabetic Kidney Disease.Diabetes. 2025 Jul 1;74(7):1205-1219. doi: 10.2337/db24-0121. Diabetes. 2025. PMID: 40183805
-
Environmental enrichment attenuates social isolation-exacerbated postoperative cognitive dysfunction in aged mice via inhibition of RAGE-HMGB1 proinflammatory signaling.Brain Res Bull. 2025 Sep;229:111462. doi: 10.1016/j.brainresbull.2025.111462. Epub 2025 Jul 11. Brain Res Bull. 2025. PMID: 40653061
-
Clinical Implications of High-mobility Group Box-1 (HMGB1) and the Receptor for Advanced Glycation End-products (RAGE) in Cutaneous Malignancy: A Systematic Review.Anticancer Res. 2017 Jan;37(1):1-7. doi: 10.21873/anticanres.11282. Anticancer Res. 2017. PMID: 28011467
-
Interventions for preventing and ameliorating cognitive deficits in adults treated with cranial irradiation.Cochrane Database Syst Rev. 2022 Nov 25;11(11):CD011335. doi: 10.1002/14651858.CD011335.pub3. Cochrane Database Syst Rev. 2022. PMID: 36427235 Free PMC article.
References
-
- Cheng AYY, Gomes MB, Kalra S, Kengne AP, Mathieu C, Shaw JE. Applying the WHO global targets for diabetes mellitus. Nat Rev Endocrinol. 2023;19:194–200. - PubMed
-
- Nagayach A, Bhaskar R, Ghosh S, Singh KK, Han SS, Sinha JK. Advancing the Understanding of diabetic encephalopathy through unravelling pathogenesis and exploring future treatment perspectives. Ageing Res Rev. 2024;100:102450. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
