

Nuclear keratin 6A upregulates human papillomavirus oncogene expression through TEAD1 interaction
- PMID: 40544293
- PMCID: PMC12181915
- DOI: 10.1186/s12985-025-02832-5
Nuclear keratin 6A upregulates human papillomavirus oncogene expression through TEAD1 interaction
Abstract
Background: Human papillomavirus (HPV) oncogenes, E6 and E7, play a critical role in cervical carcinogenesis, and their expression is regulated by cellular transcription factors bound to the long control region (LCR) in the HPV genome. To elucidate the mechanisms of HPV-induced carcinogenesis, it is important to identify host factors that control the LCR in cervical cancer cells. We previously reported that the LCR-bound transcription factor TEAD1 is critical for HPV oncogene expression. Here, we aimed to elucidate the role of stress-responsive keratin 6A (K6A), a potential cofactor for TEAD1, in HPV oncogene expression.
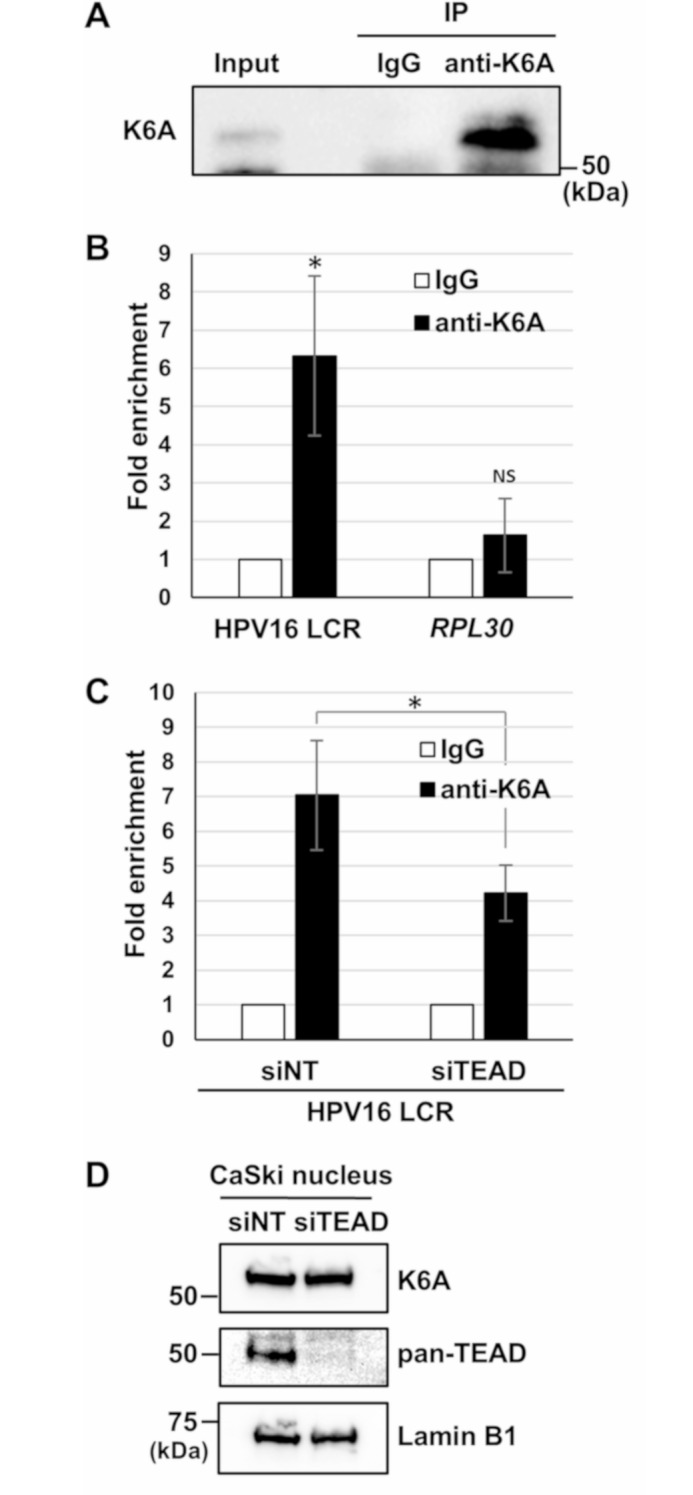
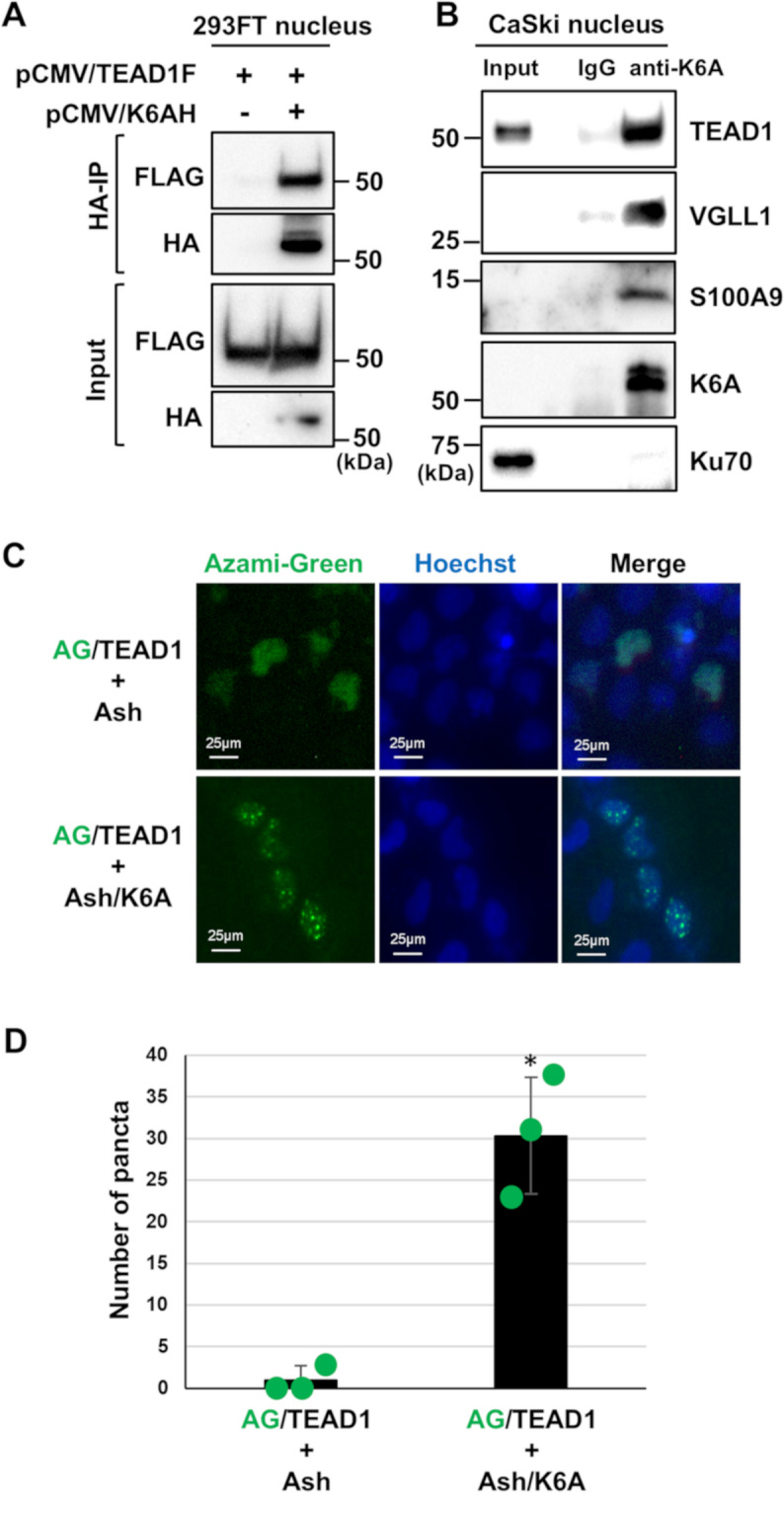
Methods: HPV16-positive cervical cancer cells were transfected with small interfering RNA (siRNA) or infected with a lentivirus expressing short hairpin RNA (shRNA) to downregulate the expression of K6A. HPV16 oncogene expression was examined by reverse transcription-quantitative polymerase chain reaction or Western blotting. Retroviral transduction was used to rescue the expression of K6A in K6A-depleted cells. Subcellular localization of K6A was analyzed by cellular fractionation followed by Western blotting. Chromatin immunoprecipitation (ChIP) assay was used to evaluate in vivo binding of K6A to the LCR. The physical interaction between K6A and TEAD1 was assessed by co-immunoprecipitation and fluorescence-based in vivo interaction assays.
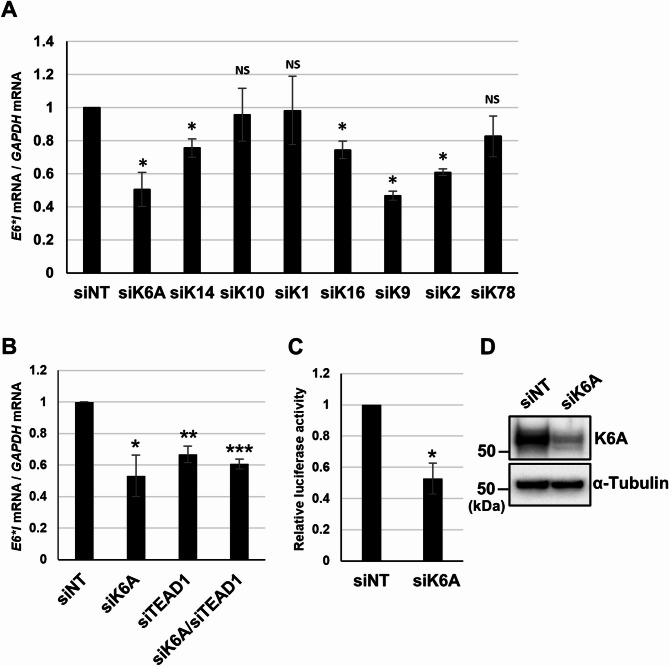
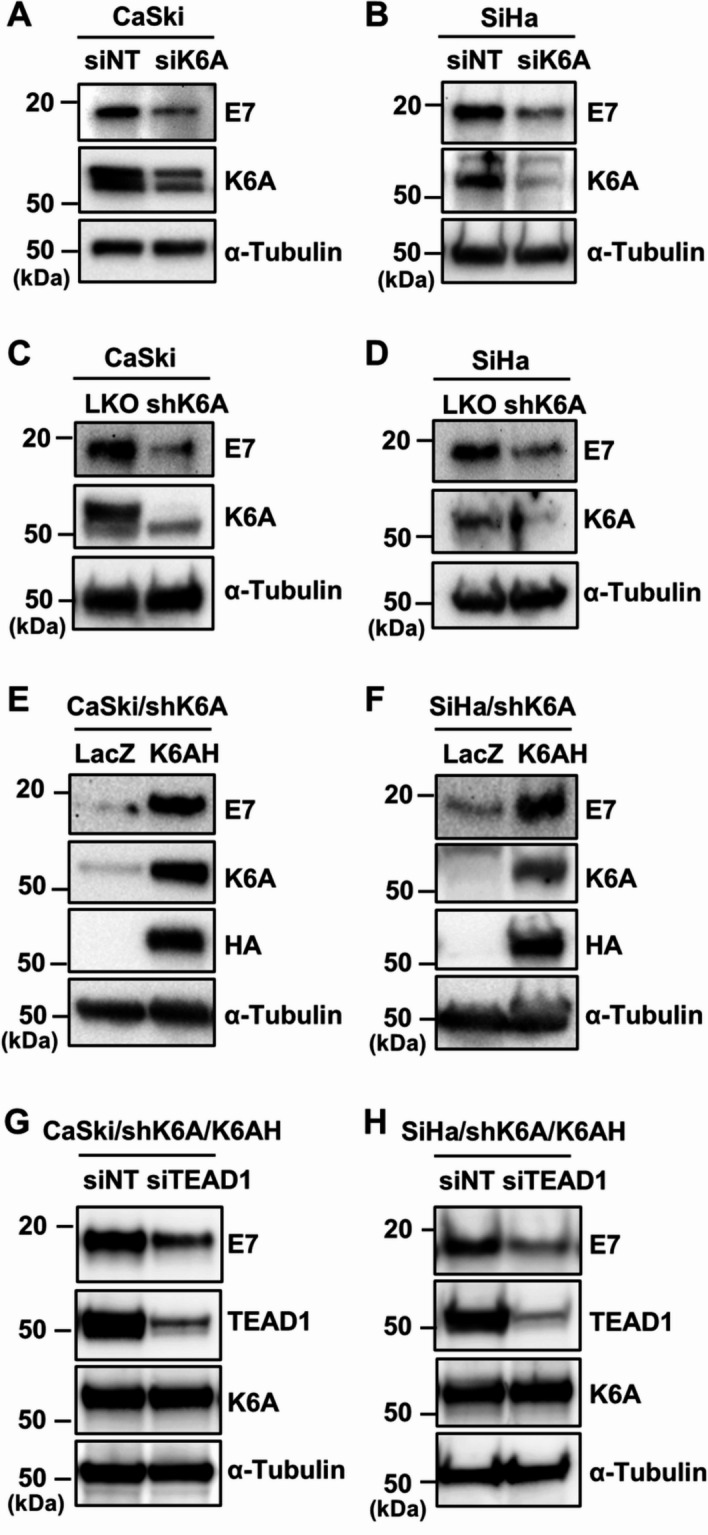
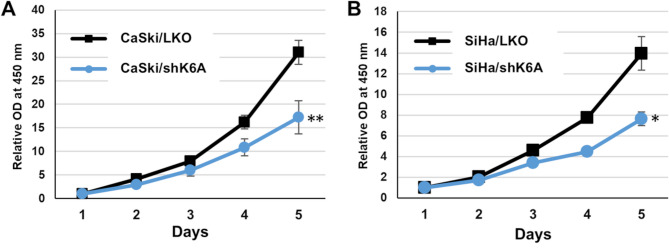
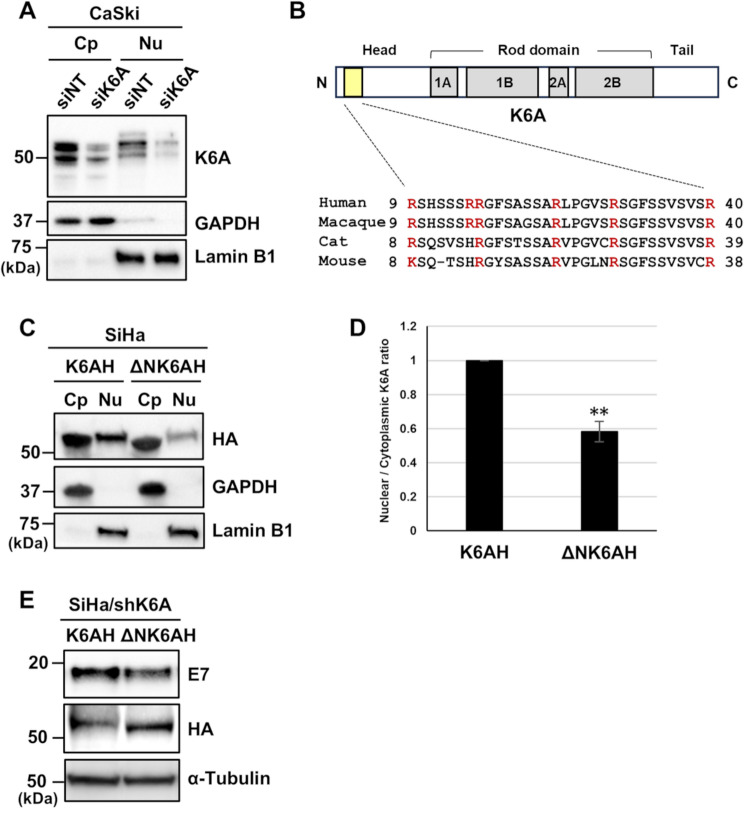
Results: Transfection of siRNA against K6A decreased levels of HPV16 E6*I mRNA and E7 protein in cervical cancer cells. Lentiviral delivery of shRNA against K6A also reduced E7 protein level and suppressed cell growth. Conversely, ectopic expression of shRNA-resistant K6A in the K6A-depleted cells restored E7 expression, and further siRNA knockdown of TEAD1 in the K6A-rescued cells attenuated E7 expression. K6A was detected in the nuclear fraction of cervical cancer cells with a functional bipartite nuclear localization signal in its N-terminus. ChIP assay showed that nuclear K6A bound to the HPV16 LCR in vivo, partially depending on the presence of the TEAD transcription factors. Finally, protein-protein interaction assays confirmed the association of K6A with TEAD1 in the nucleus.
Conclusions: Nuclear K6A associates with the LCR via TEAD1 and upregulates HPV16 oncogene expression, thereby contributing to cervical cancer development.
Supplementary Information: The online version contains supplementary material available at 10.1186/s12985-025-02832-5.
Keywords: Cervical cancer; Gene expression; Human papillomavirus; Keratin 6A; Viral oncogene.
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures
References
-
- International Agency for Research on Cancer. Cervical Cancer. https://www.iarc.who.int/cancer-type/cervical-cancer/. Accessed 28 February 2025.
Grants and funding
LinkOut - more resources
Full Text Sources
