

Enhancing Specificity, Precision, Accessibility, Flexibility, and Safety to Overcome Traditional CRISPR/Cas Editing Challenges and Shape Future Innovations
- PMID: 40548648
- PMCID: PMC12302564
- DOI: 10.1002/advs.202416331
Enhancing Specificity, Precision, Accessibility, Flexibility, and Safety to Overcome Traditional CRISPR/Cas Editing Challenges and Shape Future Innovations
Abstract
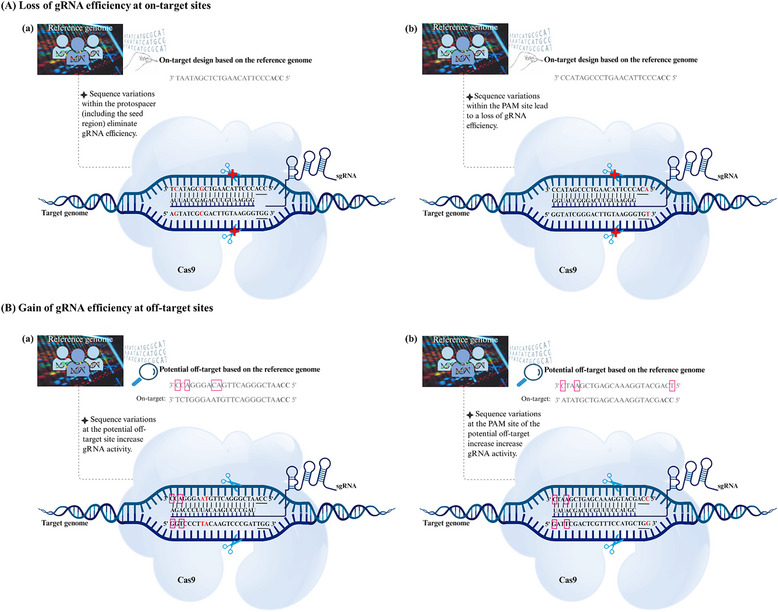
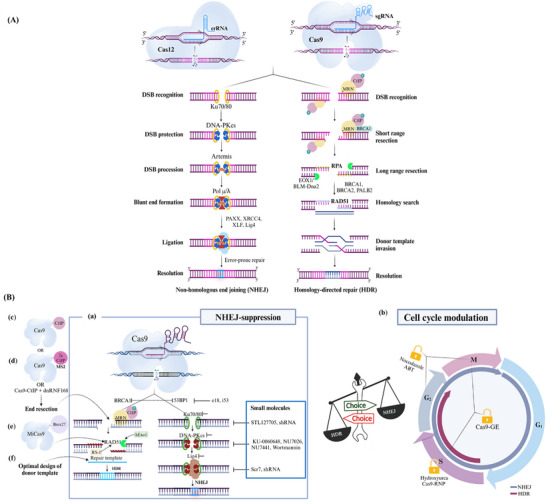
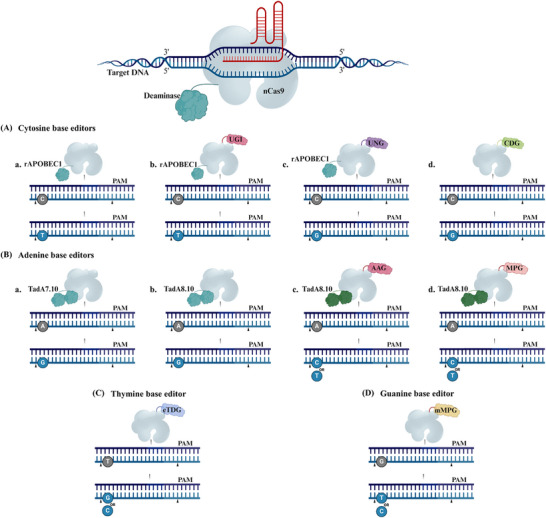
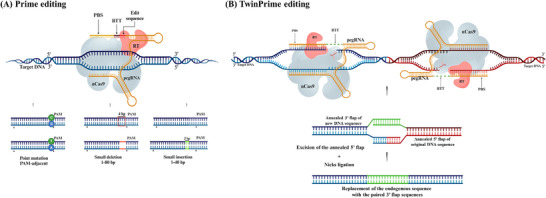
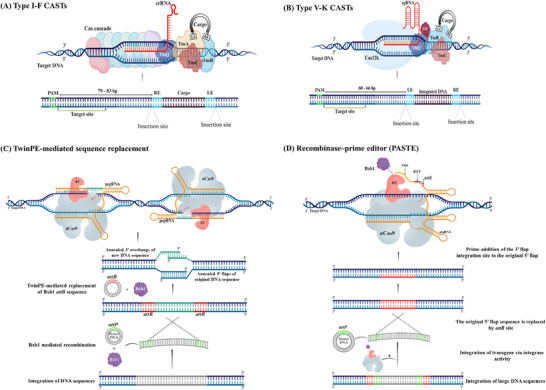
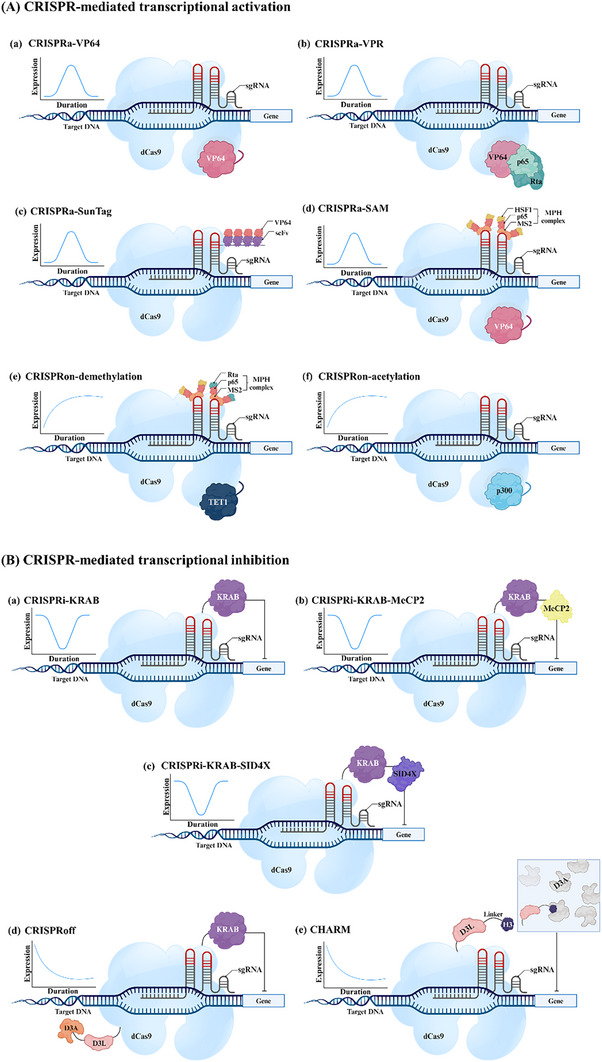
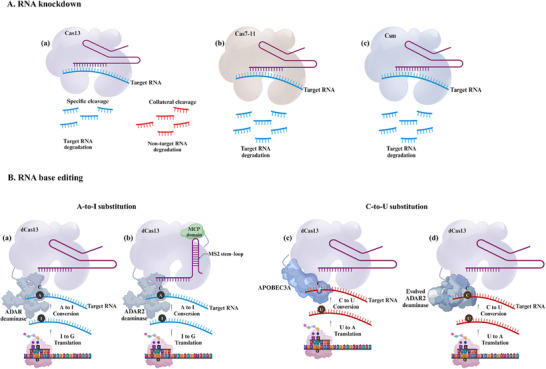
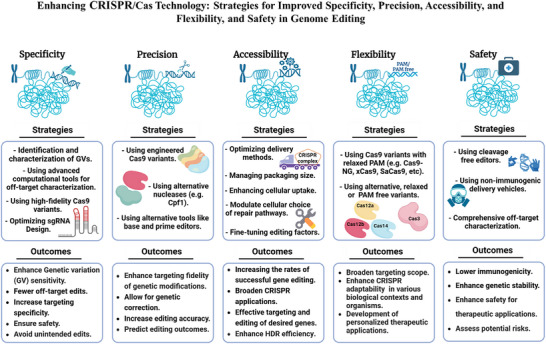
Derived from the bacterial immune system, CRISPR/Cas9 induces DSBs at specific DNA sequences, which are repaired by the cell's endogenous mechanisms, leading to gene insertions, deletions, or substitutions. Despite its transformative potential, several challenges remain in optimizing of CRISPR/Cas systems, including off-target effects, delivery methods, PAM restrictions, and the limitations of traditional editing approaches. This review focuses on the interplay between these challenges and their contributions to gene editing precision, specificity, accessibility, flexibility, and safety. How reducing off-target effects enhances specificity and safety is explored, while discussing the role of HDR-based editing in achieving precise gene modifications, alongside alternative methods such as base editing and prime editing. Improved delivery mechanisms are examined for their impact on accessibility and efficiency, while the reduction of PAM restrictions is highlighted for its contributions to flexibility. Lastly, emerging cleavage-free editing technologies are evaluated as they relate to safety and accessibility. This focused review aims to clarify the connections among these aspects and outline future research directions for advancing CRISPR-based applications.
Keywords: CRISPR/Cas; HDR; delivery systems; double‐stranded breaks; genome editing; off‐target.
© 2025 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Unlocking the potential of CRISPR-Cas9 for cystic fibrosis: A systematic literature review.Gene. 2025 Mar 20;942:149257. doi: 10.1016/j.gene.2025.149257. Epub 2025 Jan 18. Gene. 2025. PMID: 39832688
-
CRISPR/Cas genome editing in soybean: challenges and new insights to overcome existing bottlenecks.J Adv Res. 2025 Jul;73:53-72. doi: 10.1016/j.jare.2024.08.024. Epub 2024 Aug 18. J Adv Res. 2025. PMID: 39163906 Free PMC article. Review.
-
Evolution of Prime Editing Systems: Move Forward to the Treatment of Hereditary Diseases.Curr Gene Ther. 2025;25(1):46-61. doi: 10.2174/0115665232295117240405070809. Curr Gene Ther. 2025. PMID: 38623982 Review.
-
Advancing crop disease resistance through genome editing: a promising approach for enhancing agricultural production.Front Genome Ed. 2024 Jun 26;6:1399051. doi: 10.3389/fgeed.2024.1399051. eCollection 2024. Front Genome Ed. 2024. PMID: 38988891 Free PMC article. Review.
-
Nanotechnology-Based Delivery of CRISPR/Cas9 for Cancer Treatment: A Comprehensive Review.Cells. 2025 Jul 23;14(15):1136. doi: 10.3390/cells14151136. Cells. 2025. PMID: 40801569 Free PMC article. Review.
References
-
- Whitworth K., Rowland R., Ewen C., Trible B., Kerrigan M., Cino‐Ozuna A., Samuel M., Lightner J., McLaren D., Mileham A., Nat. Biotechnol. 2016, 34, 20. - PubMed
-
- Ohama M., Washio Y., Kishimoto K., Kinoshita M., Kato K., Aquac. 2020, 529, 735672.
-
- Kishimoto K., Washio Y., Yoshiura Y., Toyoda A., Ueno T., Fukuyama H., Kato K., Kinoshita M., Aquac. 2018, 495, 415.
Publication types
MeSH terms
Grants and funding
- 2023ZD04074/Biological Breeding-Major Projects
- 32325039/National Natural Science Fund of China for Distinguished Young Scholars
- 2021hszd013/Hubei Hongshan Laboratory
- W2433067/National Natural Science Foundation of China for International Young Scientists
- 32272128/National Natural Science Foundation of China
LinkOut - more resources
Full Text Sources
Miscellaneous