

Integrating taxonomic and phenotypic information through FISH-enhanced flow cytometry for microbial community dynamics analysis
- PMID: 40548738
- PMCID: PMC12323603
- DOI: 10.1128/spectrum.01973-24
Integrating taxonomic and phenotypic information through FISH-enhanced flow cytometry for microbial community dynamics analysis
Abstract
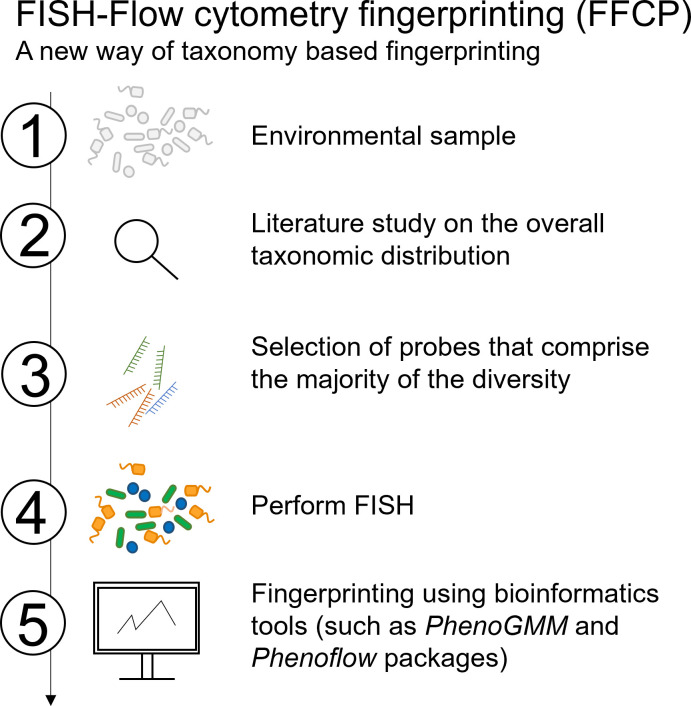
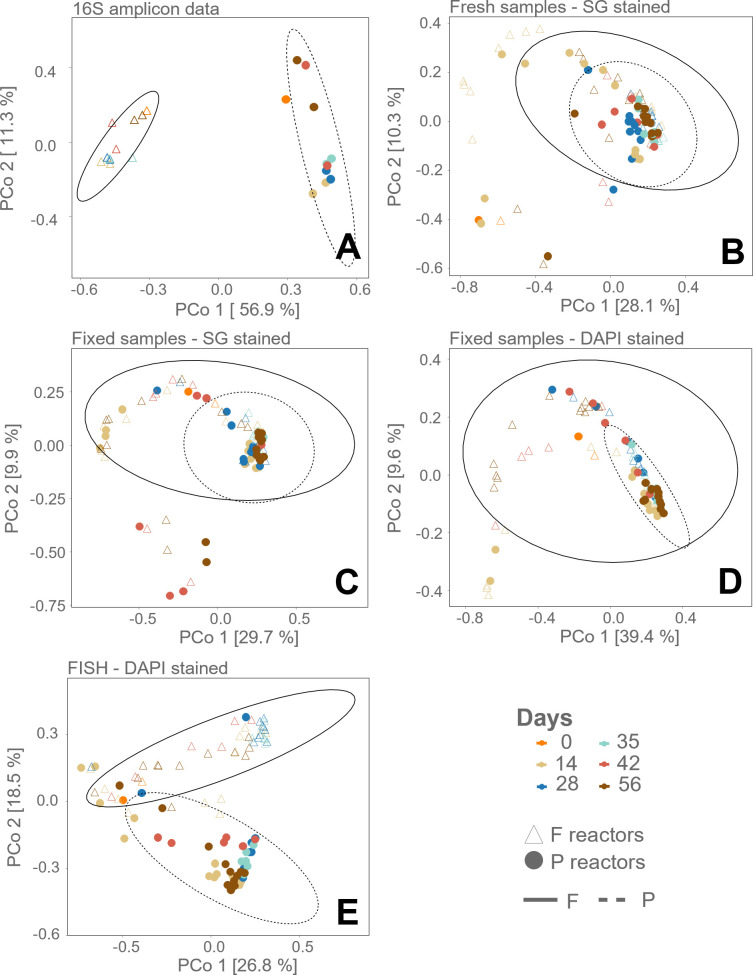
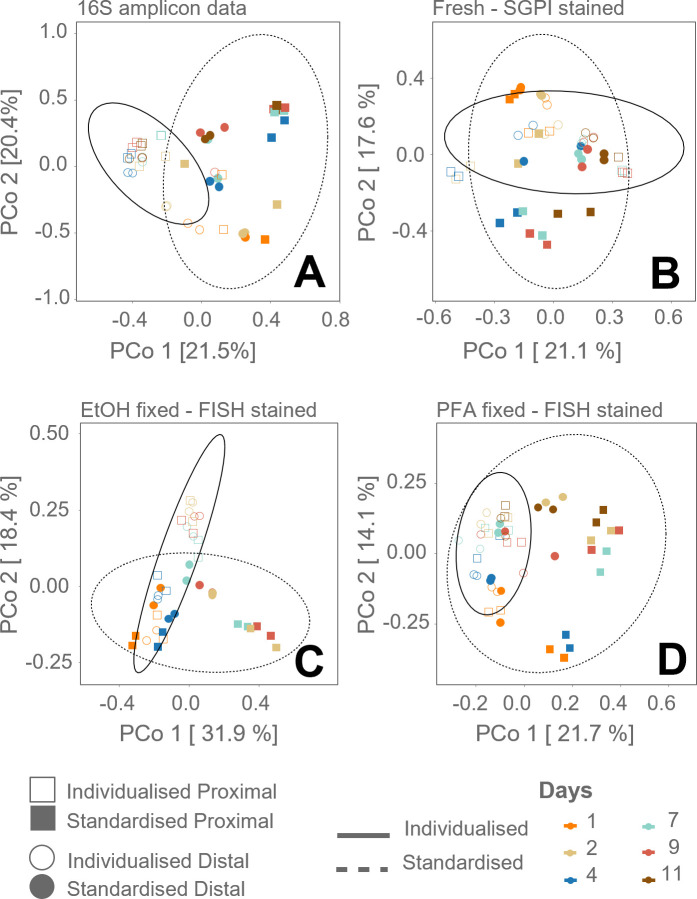
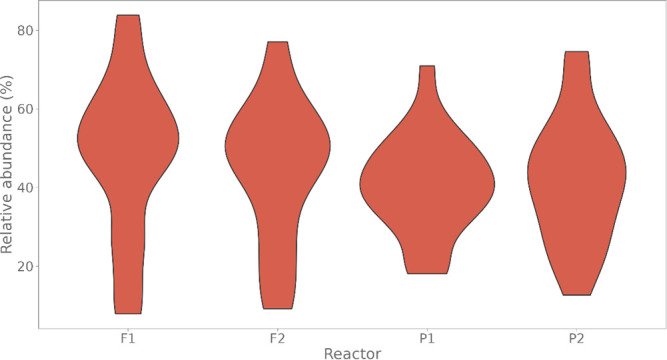
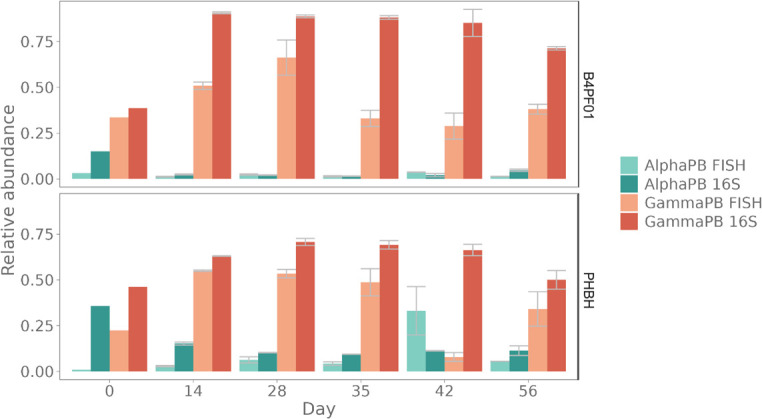
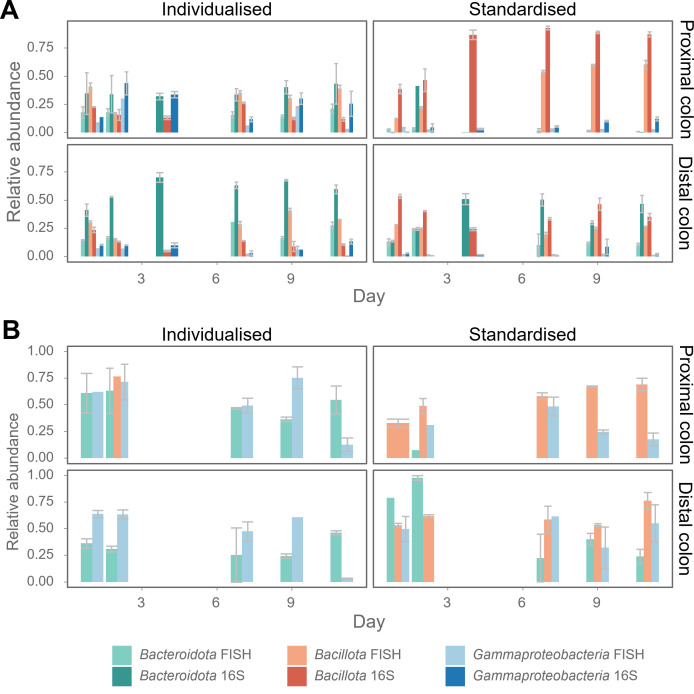
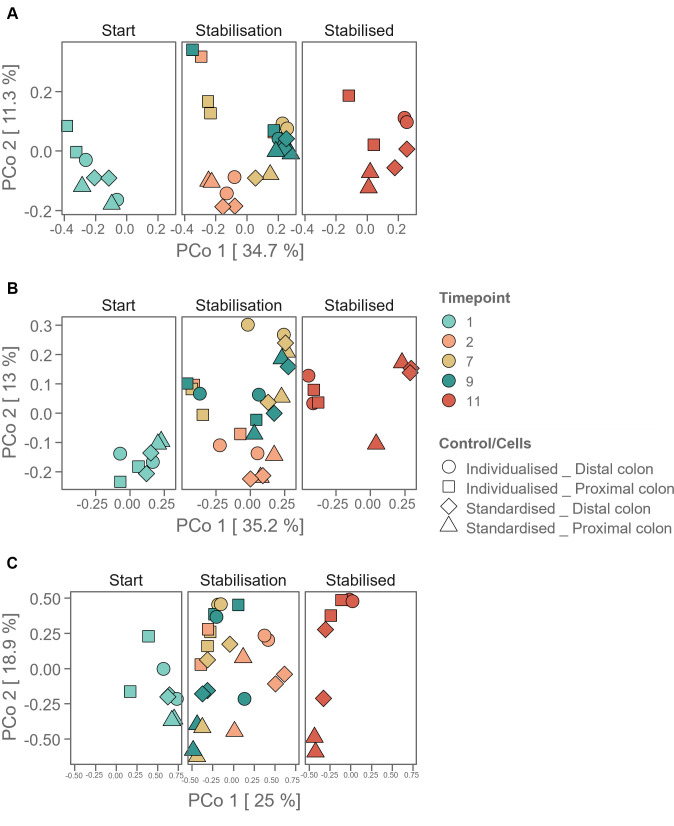
Flow cytometry is a powerful tool to monitor microbial communities, as it allows tracking both changes in the subpopulations and cell numbers at high throughput and a low sample cost. This information can be combined in a phenotypic fingerprint that can be leveraged for diversity analysis. However, as isogenic individuals can manifest phenotypic diversity, for example, due to differing physiological state and phenotypic plasticity, combining the phenotypic information with taxonomic information adds an extra dimension for describing the dynamics of a microbial community. In this research, taxonomic information was incorporated in the microbial fingerprint through fluorescent in situ hybridization (FISH) at a single-cell level. To validate this concept and explore its versatility, two ecosystems with different micro-biodiversity were considered. In the first environment, marine bacteria were monitored for plastic biodegradation in a trickling filter, and in the second, an in vitro simulated human gut microbiome was followed over time. Samples were prepared using different (staining) methods, including FISH, and beta diversity analysis was used to evaluate the level of distinction between differently treated groups in both environments. As a reference to correlate increased distinction with the incorporation of taxonomic information, 16S rRNA gene sequencing was used. Finally, a predictive algorithm was trained to correctly classify samples in the differently treated groups. The results showed that the implementation of FISH in flow cytometry provides more information on a single-cell level to answer specific scientific questions, like distinguishing between phenotypically similar communities or following a specific taxonomic group over time.
Importance: Understanding microbial communities is crucial for elucidating their role in maintaining ecosystem health and stability. Researchers are increasingly interested in studying microbial communities by looking at not just their genetic makeup but also their physical traits and functions. In our study, we used common techniques like fluorescence in situ hybridization and flow cytometry, along with advanced data analysis, to better understand these communities. This combination allowed us to gather and use data more effectively, demonstrating that these easy-to-use methods, when paired with proper analysis, can enhance our understanding of changing microbial ecosystems.
Keywords: flow cytometry; fluorescence in situ hybridization; microbial community dynamics; phenotypic fingerprinting.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Props R, Monsieurs P, Mysara M, Clement L, Boon N. 2016. Measuring the biodiversity of microbial communities by flow cytometry. Methods Ecol Evol 7:1376–1385. doi: 10.1111/2041-210X.12607 - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
