

Serotonin receptor 5-HT2A as a potential target for HCC immunotherapy
- PMID: 40550560
- PMCID: PMC12186046
- DOI: 10.1136/jitc-2024-011088
Serotonin receptor 5-HT2A as a potential target for HCC immunotherapy
Abstract
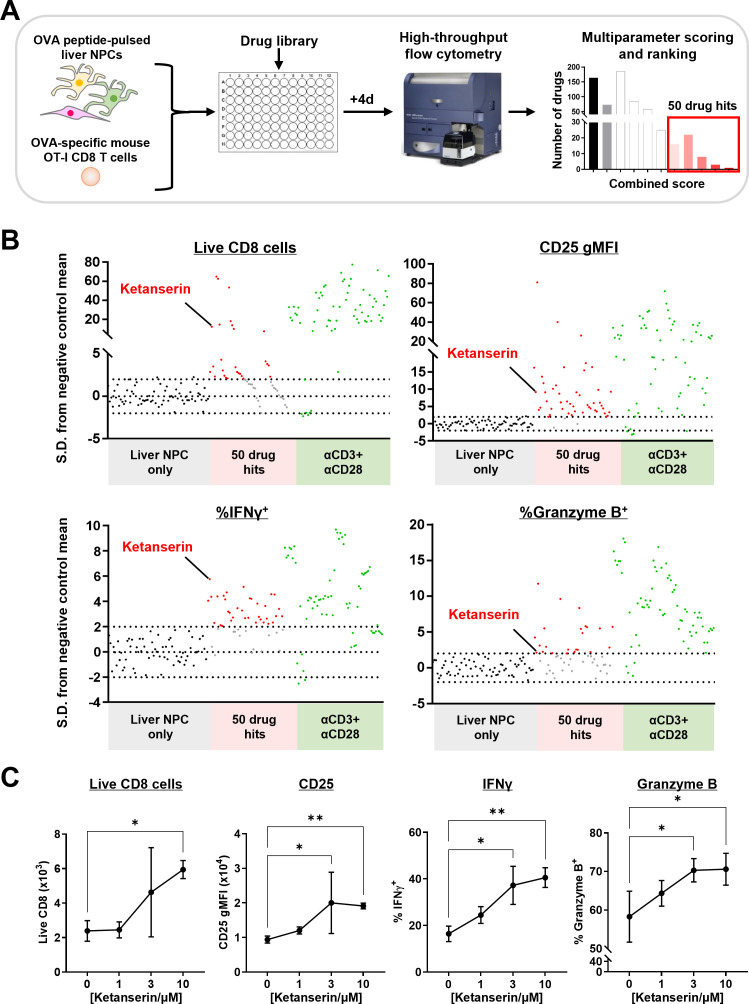
Background: While recent clinical trials of combination immunotherapies for hepatocellular carcinoma (HCC) have shown promising clinical efficacy and survival improvements breakthroughs, there is still much room for further improvement. A key limiting factor for HCC immunotherapy is the intrinsic immunosuppression within the liver microenvironment, resulting in suboptimal priming of tumor-specific CD8 cytotoxic T cells and thus immune evasion by the tumor. Hence, identifying new key molecular pathways suppressing T-cell responses within the liver is critical for the rational design of more effective combination immunotherapies for HCC.
Methods: We identified the 5-HT2A serotonin receptor as a potential target for HCC immunotherapy in a chemical screening approach and validated that targeting 5-HT2A signaling could be a viable approach for HCC immunotherapy via in vitro and in vivo studies.
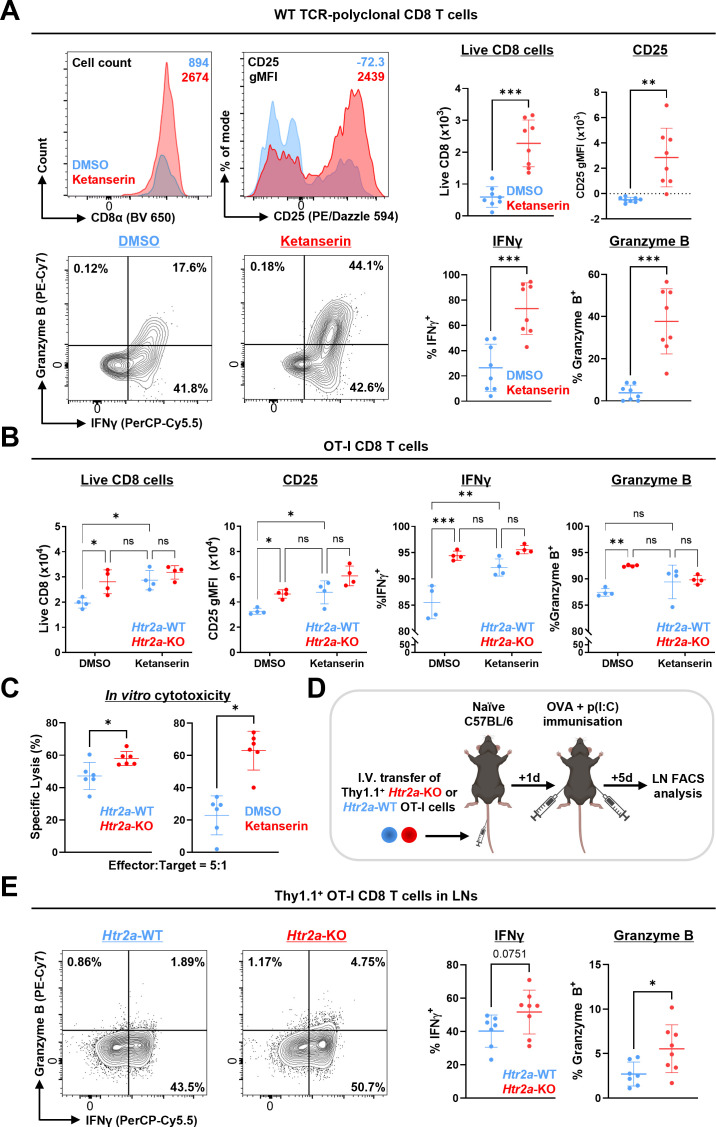
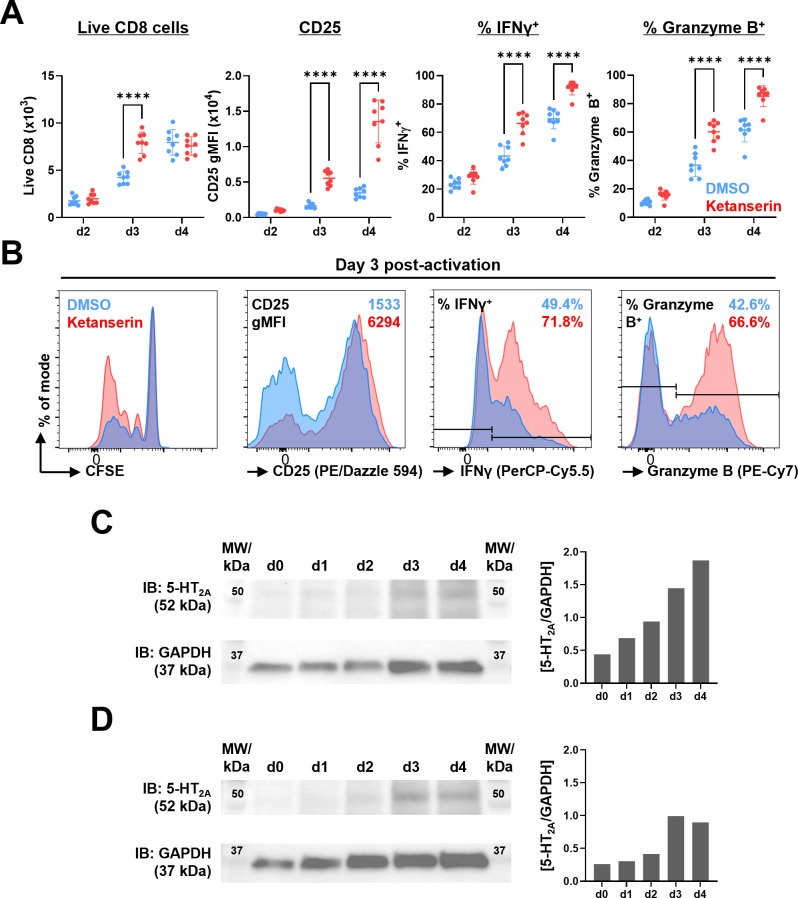
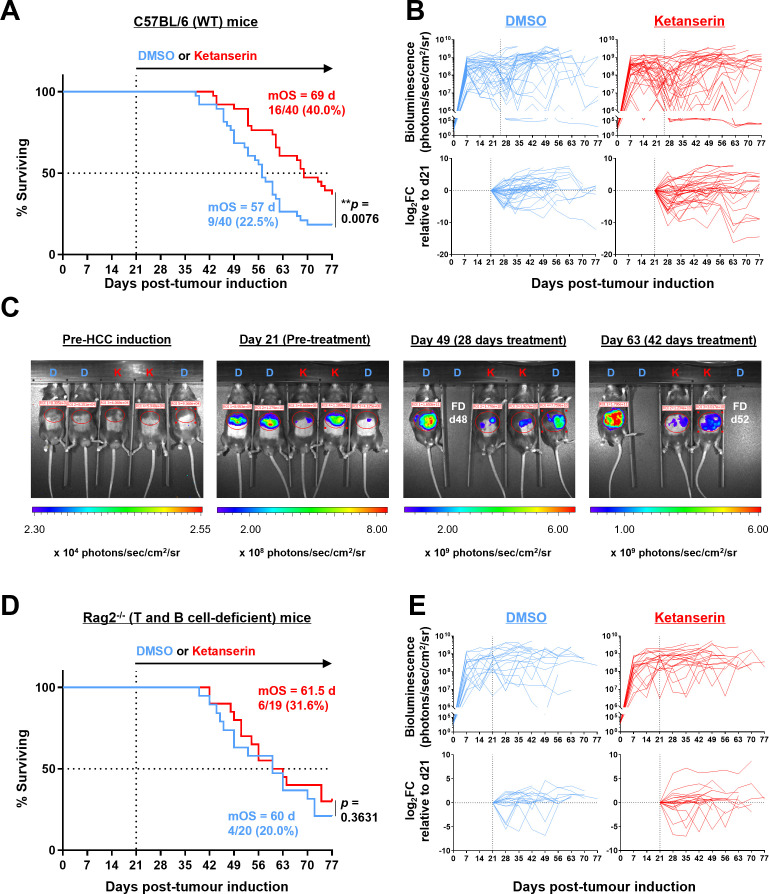
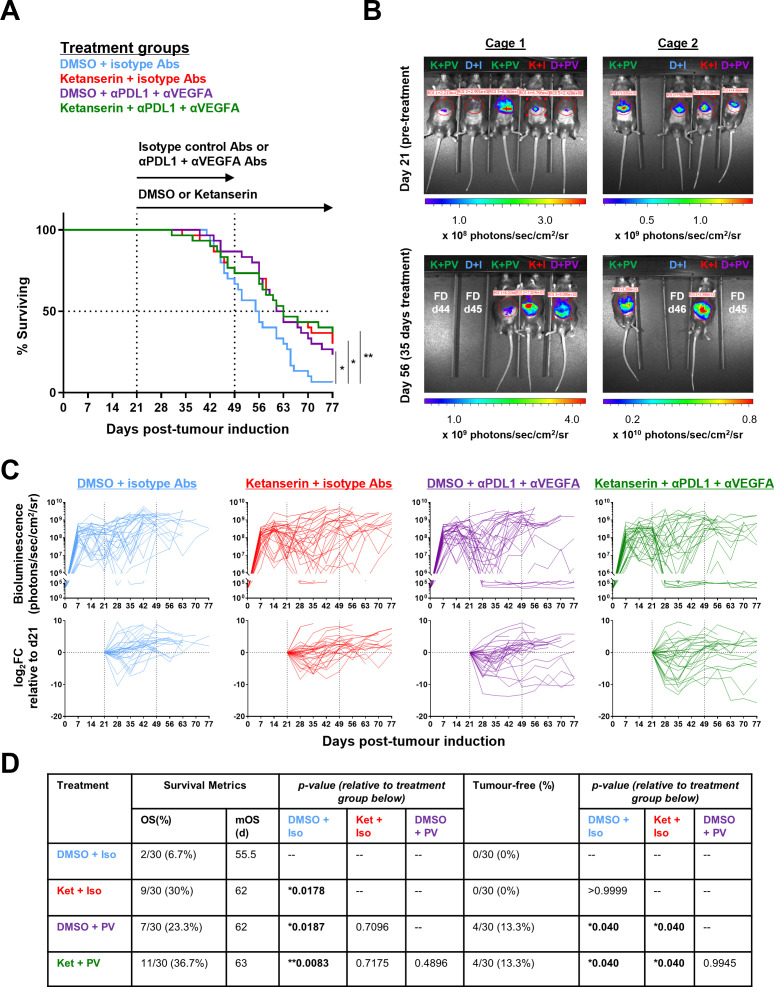
Results: Disruption of 5-HT2A signaling using either a selective antagonist small molecule, ketanserin, or by knockout of its coding gene Htr2a augments the cytotoxic effector phenotype of mouse CD8 T cells activated in vitro with immunosuppressive liver non-parenchymal cells. Ketanserin treatment of in vitro activated human CD8 T cells also increased expression of the cytotoxic effector molecules granzyme B and perforin. Abrogation of 5-HT2A signaling was associated with increased expression of cytotoxicity-related genes such as granzyme B and reduced expression of transcription factors downstream of MAP kinase signaling. In vivo, systemic ketanserin treatment significantly prolonged survival of HCC tumor-bearing mice and was non-inferior to α-programmed death ligand 1 (PD-L1)+α-vascular endothelial growth factor A (VEGFA) combination antibody treatment. Combining ketanserin with αPD-L1+αVEGFA antibodies also significantly prolonged survival relative to control-treated mice while preserving the occurrence of complete tumor regression observed with αPD-L1+αVEGFA treatment alone.
Conclusions: Together, our data describe a role for 5-HT2A as a negative regulator of the cytotoxic effector phenotype in CD8 T cells and highlight the therapeutic potential of targeting 5-HT2A for HCC immunotherapy.
Keywords: Combination therapy; Hepatocellular Carcinoma; Immunotherapy; T cell.
© Author(s) (or their employer(s)) 2025. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ Group.
Conflict of interest statement
Competing interests: None declared
Figures


References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials