

ER-induced PERK/TFEB cascade sequentially modulates mitochondrial dynamics during cranial suture expansion
- PMID: 40550802
- PMCID: PMC12185732
- DOI: 10.1038/s41413-025-00427-y
ER-induced PERK/TFEB cascade sequentially modulates mitochondrial dynamics during cranial suture expansion
Abstract
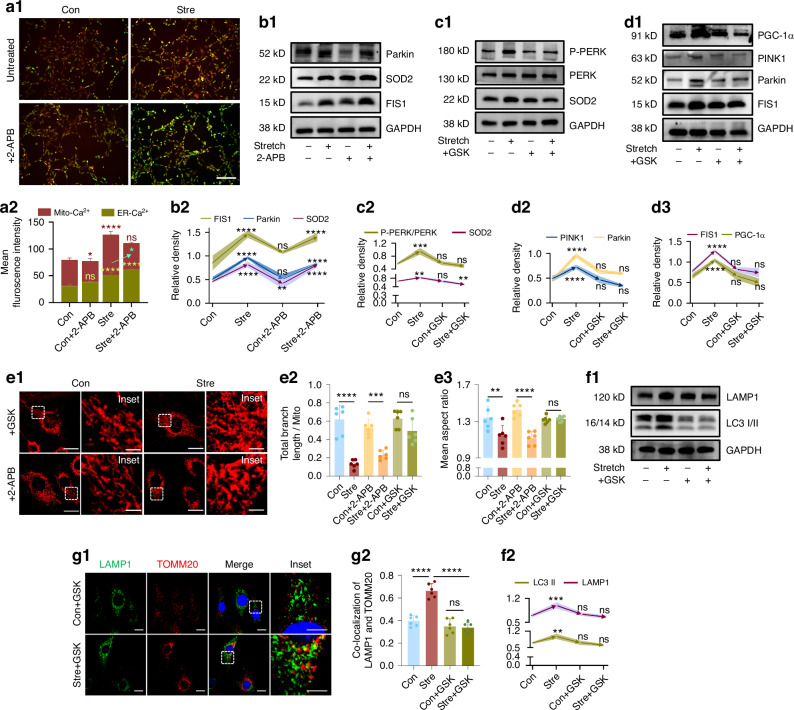
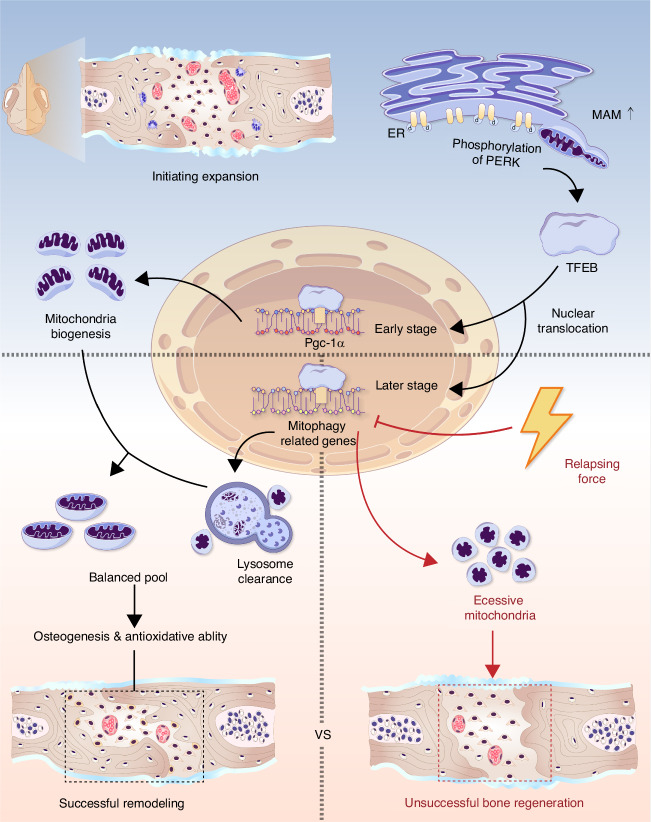
The effectiveness of cranial suture expansion therapy hinges on the timely and adequate regeneration of bone tissue in response to mechanical stimuli. To optimize clinical outcomes and prevent post-expansion relapse, we delved into the underlying mechanisms governing bone remodeling during the processes of suture expansion and relapse. Our findings revealed that in vitro stretching bolstered mesenchymal stem cells' antioxidative and osteogenic capacity by orchestrating mitochondrial activities, which governed by force-induced endoplasmic reticulum (ER) stress. Nonetheless, this signal transduction occurred through the activation of protein kinase R-like ER kinase (PERK) at the ER-mitochondria interface, rather than ER-mitochondria calcium flow as previously reported. Subsequently, PERK activation triggered TFEB translocation to the nucleus, thus regulating mitochondrial dynamics transcriptionally. Assessment of the mitochondrial pool during expansion and relapse unveiled a sequential, two-phase regulation governed by the ER stress/p-PERK/TFEB signaling cascade. Initially, PERK activation facilitated TFEB nuclear localization, stimulating mitochondrial biogenesis through PGC1-α, thereby addressing energy demands during the initial phase. Subsequently, TFEB shifted focus towards ensuring adequate mitophagy for mitochondrial quality maintenance during the remodeling process. Premature withdrawal of expanding force disrupted this sequential regulation, leading to compromised mitophagy and the accumulation of dysfunctional mitochondria, culminating in suboptimal bone regeneration and relapse. Notably, pharmacological activation of mitophagy effectively mitigated relapse and attenuated bone loss, while its inhibition impeded anticipated bone growth in remodeling progress. Conclusively, we elucidated the ER stress/p-PERK/TFEB signaling orchestrated sequential mitochondria biogenesis and mitophagy under mechanical stretch, thus ensuring antioxidative capacity and osteogenic potential of cranial suture tissues.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures







References
MeSH terms
Substances
Grants and funding
- 82370988/National Natural Science Foundation of China (National Science Foundation of China)
- 32271416/National Natural Science Foundation of China (National Science Foundation of China)
- 81870743/National Natural Science Foundation of China (National Science Foundation of China)
- 82170934/National Natural Science Foundation of China (National Science Foundation of China)
- 2024YFHZ0043/Sichuan Provincial Department of Science and Technology | Sichuan Province Science and Technology Support Program
LinkOut - more resources
Full Text Sources
