

Using large-scale population-based data to improve disease risk assessment of clinical variants
- PMID: 40551016
- PMCID: PMC12321300
- DOI: 10.1038/s41588-025-02212-3
Using large-scale population-based data to improve disease risk assessment of clinical variants
Abstract
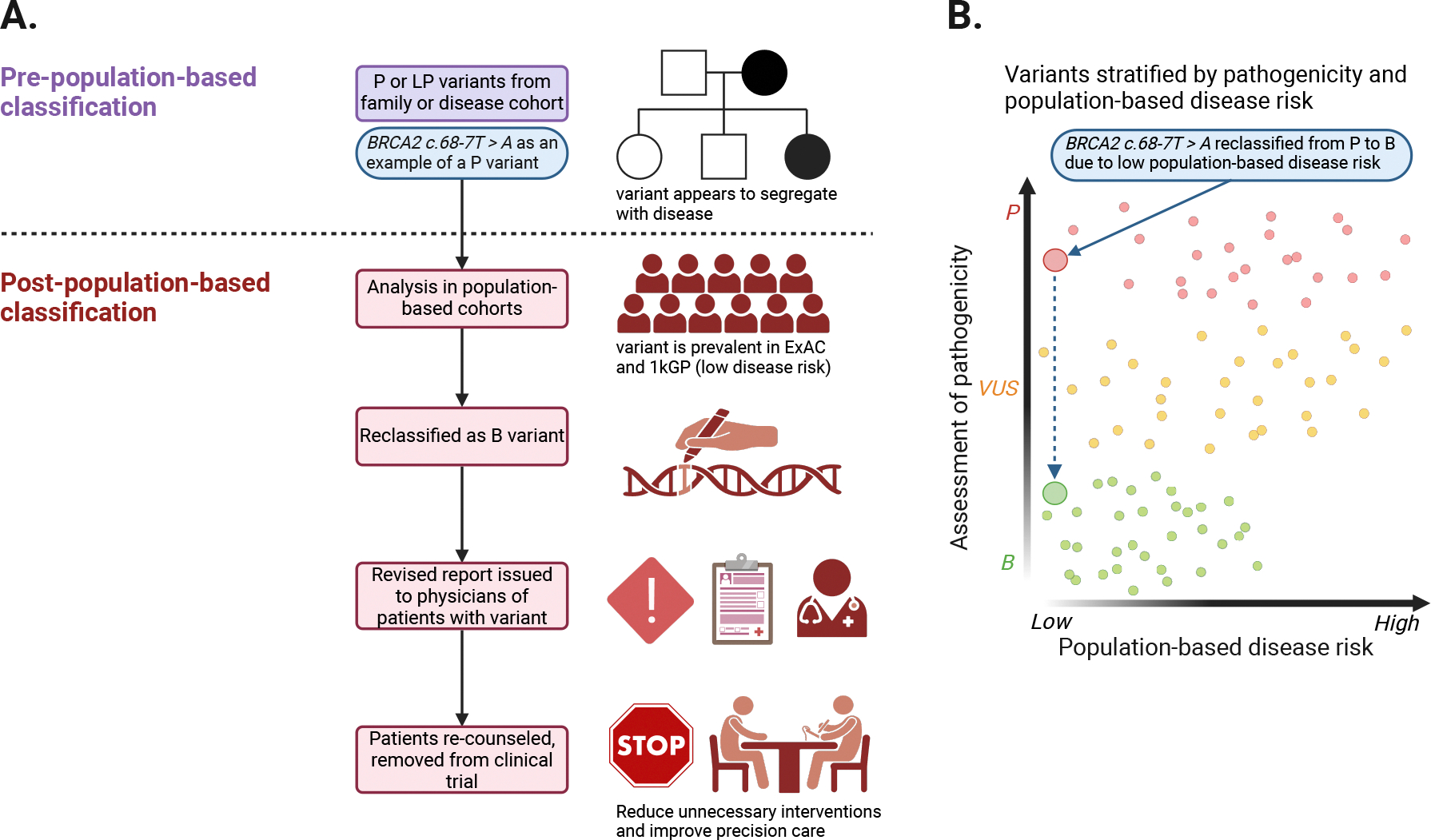
Understanding the disease risk of genetic variants is fundamental to precision medicine. Estimates of penetrance-the probability of disease for individuals with a variant allele-rely on disease-specific cohorts, clinical testing and emerging electronic health record (EHR)-linked biobanks. These data sources, while valuable, each have limitations in quality, representativeness and analyzability. Here, we provide a historical account of the currently accepted pathogenicity classification system and data available in ClinVar, a public archive that aggregates variant interpretations but lacks detailed data for accurate penetrance assessment, highlighting its oversimplification of disease risk. We propose an integrative Bayesian framework that unifies pathogenicity and penetrance, leveraging both functional and real-world evidence to refine risk predictions. In addition, we advocate for enhancing ClinVar with the inclusion of high-priority phenotypes, age-stratified data and population-based cohorts linked to EHRs. We suggest developing a community repository of population-based penetrance estimates to support the clinical application of genetic data.
© 2025. Springer Nature America, Inc.
Conflict of interest statement
Competing interests: R.D. reported being a scientific cofounder, consultant and equity holder for Pensieve Health (pending) and a consultant for Variant Bio and Character Bio. J.M.E. reported being a cofounder, board member and executive of the nonprofit Center for Genomic Interpretation, with part of its mission overlapping with the interests of this work, specifically the mission to encourage careful stewardship of clinical genetics. J.M.E. is also the founder of and a consultant for Grandview Consulting LLC, not related to this work. K.-L.H. is a founder of Open Box Science, not related to this work. W.K.C. is on the Board of Directors of Prime Medicine and Rallybio, not related to this work. The other authors declare no competing interests.
Figures
Similar articles
-
Quality improvement strategies for diabetes care: Effects on outcomes for adults living with diabetes.Cochrane Database Syst Rev. 2023 May 31;5(5):CD014513. doi: 10.1002/14651858.CD014513. Cochrane Database Syst Rev. 2023. PMID: 37254718 Free PMC article.
-
Finding buried genetic test results in the electronic health record is inefficient and variable across institutions.Ther Adv Rare Dis. 2025 Jul 11;6:26330040251356521. doi: 10.1177/26330040251356521. eCollection 2025 Jan-Dec. Ther Adv Rare Dis. 2025. PMID: 40657271 Free PMC article.
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
Cost-effectiveness of using prognostic information to select women with breast cancer for adjuvant systemic therapy.Health Technol Assess. 2006 Sep;10(34):iii-iv, ix-xi, 1-204. doi: 10.3310/hta10340. Health Technol Assess. 2006. PMID: 16959170
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
References
-
- CDC. Tier 1 Genomics Applications and their Importance to Public Health. Office of Genomics and Precision Public Health https://www.cdc.gov/genomics/implementation/toolkit/tier1.htm (2014).
-
- Sturm AC et al. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 72, 662–680 (2018). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
