

Development and characterization of a low-affinity humanized CD19 chimeric antigen receptor for B-cell malignancies
- PMID: 40552142
- PMCID: PMC12182840
- DOI: 10.1016/j.bneo.2024.100048
Development and characterization of a low-affinity humanized CD19 chimeric antigen receptor for B-cell malignancies
Abstract
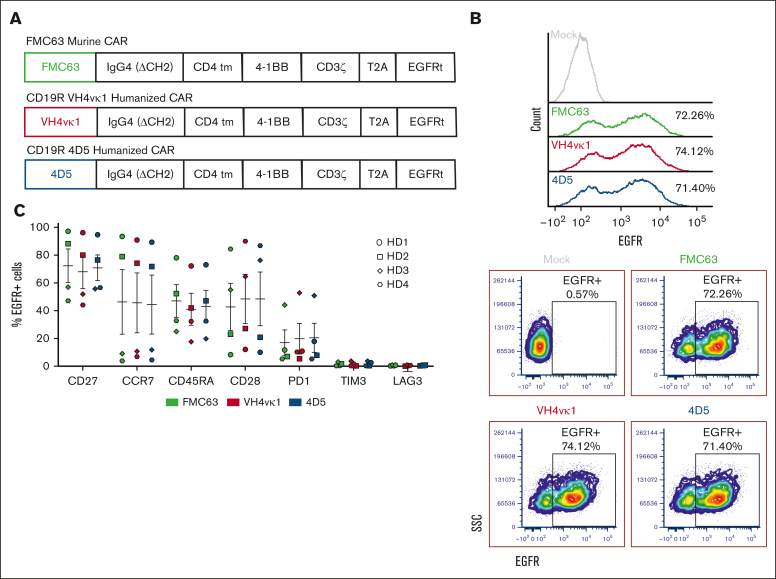
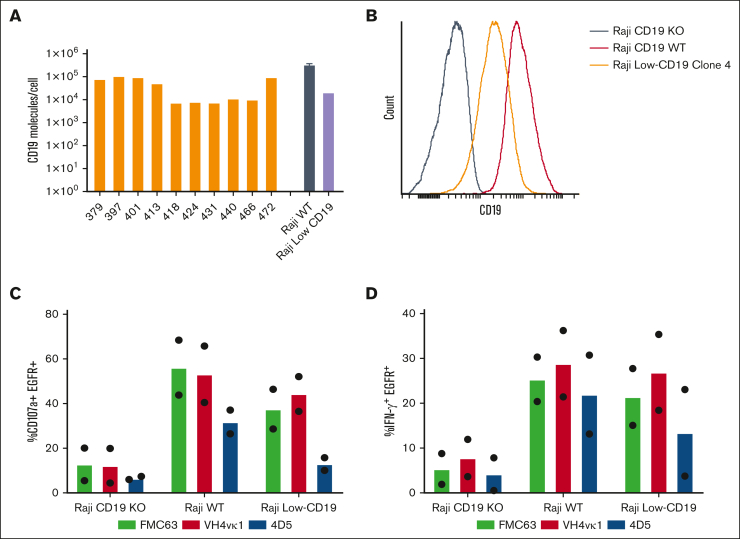
In this study, we aim to develop a humanized CD19 chimeric antigen receptor (CAR) that matches the potency of the FMC63 CAR and potentially reduces the risk of immunogenicity. The murine FMC63 single-chain variable fragment (scFv) was humanized yielding 2 lead candidate scFvs, VH4vκ1 and 4D5, which exhibit weaker binding affinity than FMC63 scFv. These humanized CD19-scFvs were incorporated into CAR constructs to generate huCD19R(VH4Vκ1) and huCD19R(4D5) CARs, both containing the 41BB costimulatory domain. The antitumor activity of the CAR T cells was assessed against CD19+ and CD19 low-expressing tumors. FMC63 CAR T cells with the same backbone in all studies were used as controls. The results showed that the huCD19R(VH4vκ1) CAR T cells exhibited similar expansion, phenotype, and effector function to the FMC63 CAR upon stimulation with CD19 targets. When the CAR T cells were challenged with CD19-bearing tumors, the huCD19R(VH4vκ1) CAR T cells showed similar proliferation to the FMC63 CAR T cells, whereas the huCD19R(4D5) CAR T cells essentially failed to proliferate. Moreover, the huCD19R(VH4vκ1) CAR T cells exhibited significantly better in vivo antitumor activity than the huCD19R(4D5) CAR T cells when tested against tumors expressing a range of CD19 antigens. Finally, using a hybrid model, we found that the huCD19R(VH4vκ1) T cells had a comparable cytokine secretion profile to that of FMC63 CAR T cells. Furthermore, the huCD19R(VH4vκ1) CAR T cells exhibited efficacy against both CD19+ and engineered CD19 low-expressing tumors. These findings suggest that huCD19R(VH4vκ1) CAR T cells may offer enhanced persistence and represent a promising candidate for clinical translation as a therapy for CD19+ tumors.
© 2024 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: L.A.S., J.C.W., S.J.F. X.W., C.E.B. are the inventors of the huCD19CAR patent application. The remaining authors declare no competing financial interests. The current affiliation for L.A.S. is the Department of Chemical, Biological, and Materials Engineering, University of South Florida, Tampa, FL
Figures






References
-
- Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–852. - PubMed
-
- Bouchkouj N, Kasamon YL, de Claro RA, et al. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res. 2019;25(6):1702–1708. - PubMed
-
- Pehlivan KC, Duncan BB, Lee DW. CAR-T cell therapy for acute lymphoblastic leukemia: transforming the treatment of relapsed and refractory disease. Curr Hematol Malig Rep. 2018;13(5):396–406. - PubMed
LinkOut - more resources
Full Text Sources
