

AAnet Resolves a Continuum of Spatially Localized Cell States to Unveil Intratumoral Heterogeneity
- PMID: 40552975
- PMCID: PMC12498099
- DOI: 10.1158/2159-8290.CD-24-0684
AAnet Resolves a Continuum of Spatially Localized Cell States to Unveil Intratumoral Heterogeneity
Abstract
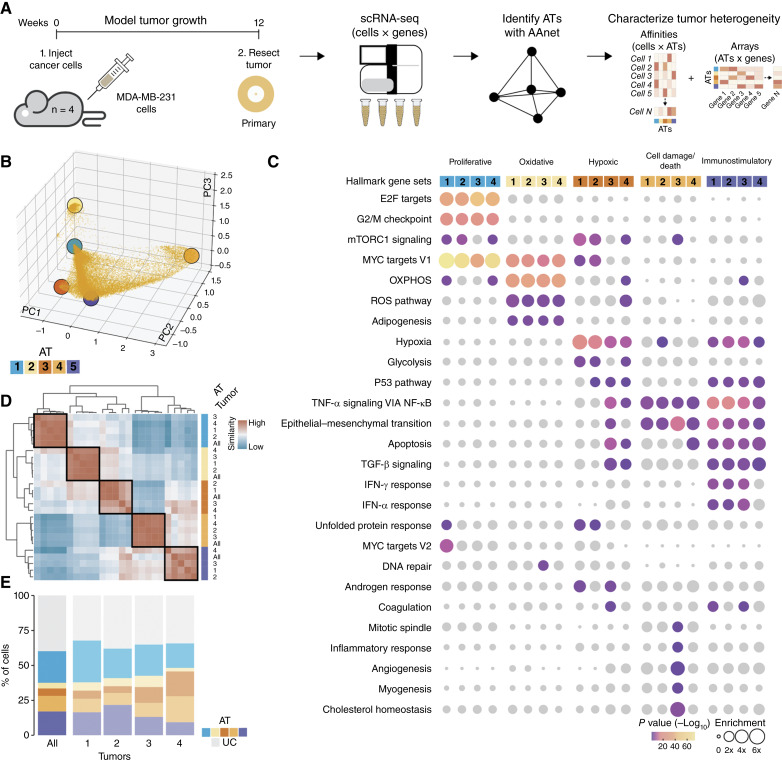
Identifying functionally important cell states and structure within heterogeneous tumors remains a significant biological and computational challenge. Current clustering- or trajectory-based models are ill-equipped to address the notion that cancer cells reside along a phenotypic continuum. We present Archetypal Analysis network (AAnet), a neural network that learns archetypal states within a phenotypic continuum in single-cell data. Unlike traditional archetypal analysis, AAnet learns archetypes (AT) in a simplex-shaped neural network latent space. Using preclinical and clinical models of breast cancer, AAnet resolves distinct cell states and processes, including cell proliferation, hypoxia, metabolism, and immune interactions. Primary tumor ATs are recapitulated in matched liver, lung, and lymph node metastases. Spatial transcriptomics reveals archetypal organization within the tumor and intra-archetypal mirroring between cancer and adjacent stromal cells. AAnet identifies GLUT3 within the hypoxic AT that proves critical for tumor growth and metastasis. AAnet is a powerful tool, capturing complex, functional cell states from multimodal data.
Significance: Defining critical cell states among cells that reside along a phenotypic continuum is a current biological and computational challenge. In this study, we present AAnet, a neural network that learns archetypal cell states of cancer cells. AAnet defines discrete spatially localized ATs that resolve intratumoral heterogeneity.
©2025 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
S.E. Youlten reports personal fees from Compbiosphere outside the submitted work, as well as being the founder and director of Compbiosphere, a computational biology analytics and consulting company. D.B. Burkhardt reports other support from Cellarity, Inc. and NVIDIA and grants from Chan Zuckerberg Initiative outside the submitted work. A. Benz reports other support from Cellarity, Inc., outside the submitted work. J. Lundeberg reports grants from The Swedish Cancer Society during the conduct of the study, as well as personal fees from 10× Genomics, Inc. outside the submitted work. S. Krishnaswamy reports personal fees from Meta and Latent Alpha outside the submitted work. No disclosures were reported by the other authors.
Figures






References
-
- McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 2017;168:613–28. - PubMed
MeSH terms
Grants and funding
- GNT1184009/National Health and Medical Research Council (NHMRC)
- GNT2012848/National Health and Medical Research Council (NHMRC)
- GNT2028506/National Health and Medical Research Council (NHMRC)
- GNT2019437/National Health and Medical Research Council (NHMRC)
- GNT2033083/National Health and Medical Research Council (NHMRC)
LinkOut - more resources
Full Text Sources
Medical
