

Infectome analysis of bat kidneys from Yunnan province, China, reveals novel henipaviruses related to Hendra and Nipah viruses and prevalent bacterial and eukaryotic microbes
- PMID: 40554741
- PMCID: PMC12187171
- DOI: 10.1371/journal.ppat.1013235
Infectome analysis of bat kidneys from Yunnan province, China, reveals novel henipaviruses related to Hendra and Nipah viruses and prevalent bacterial and eukaryotic microbes
Abstract
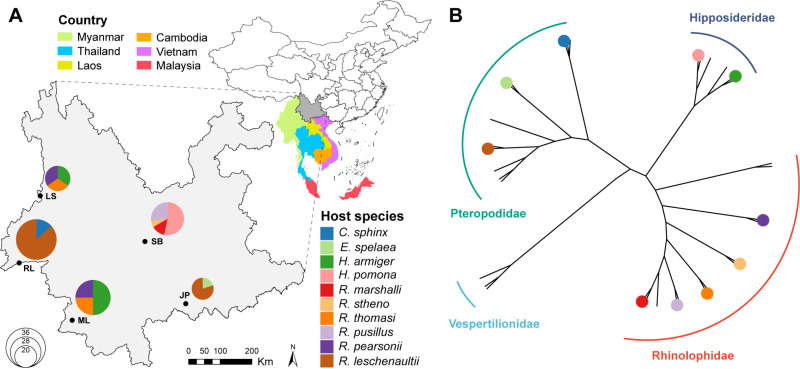
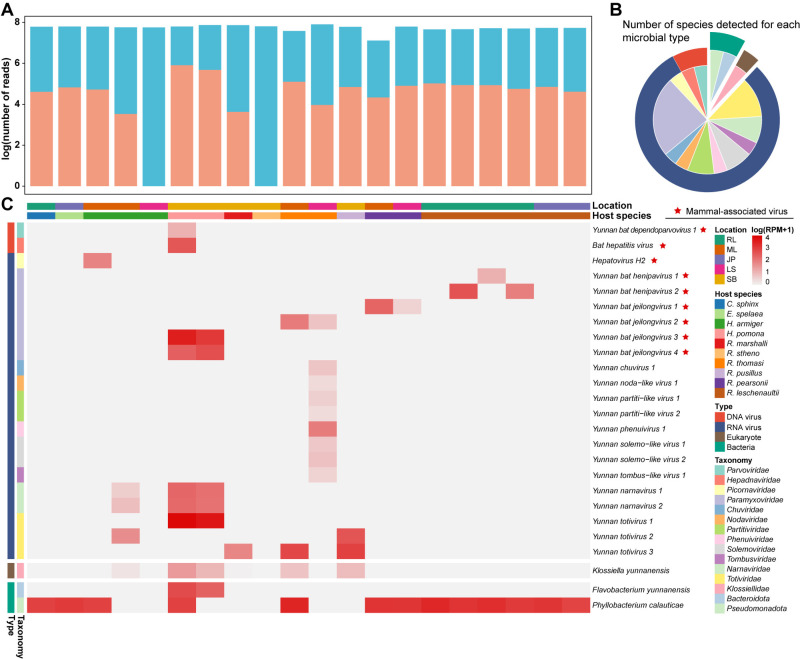
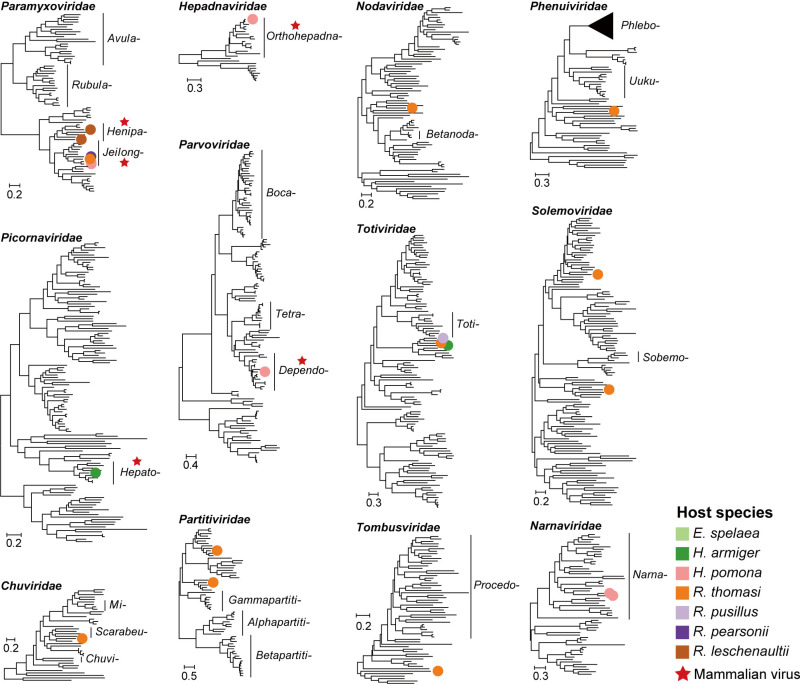
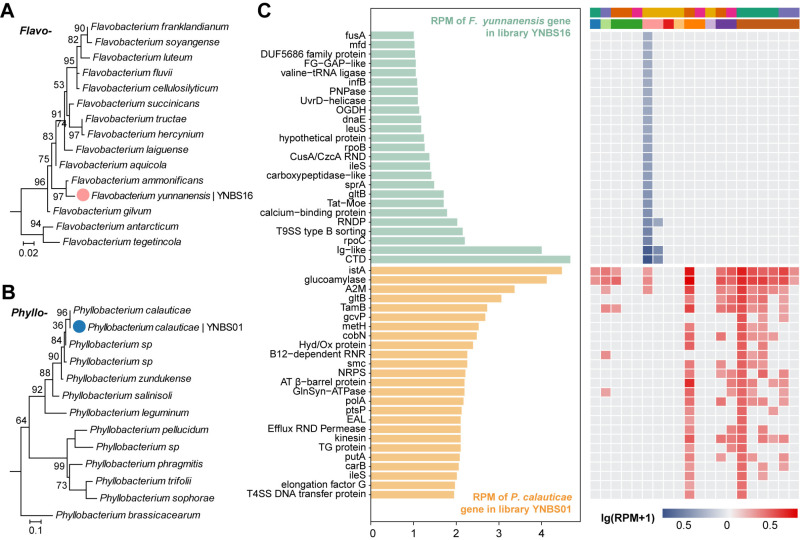
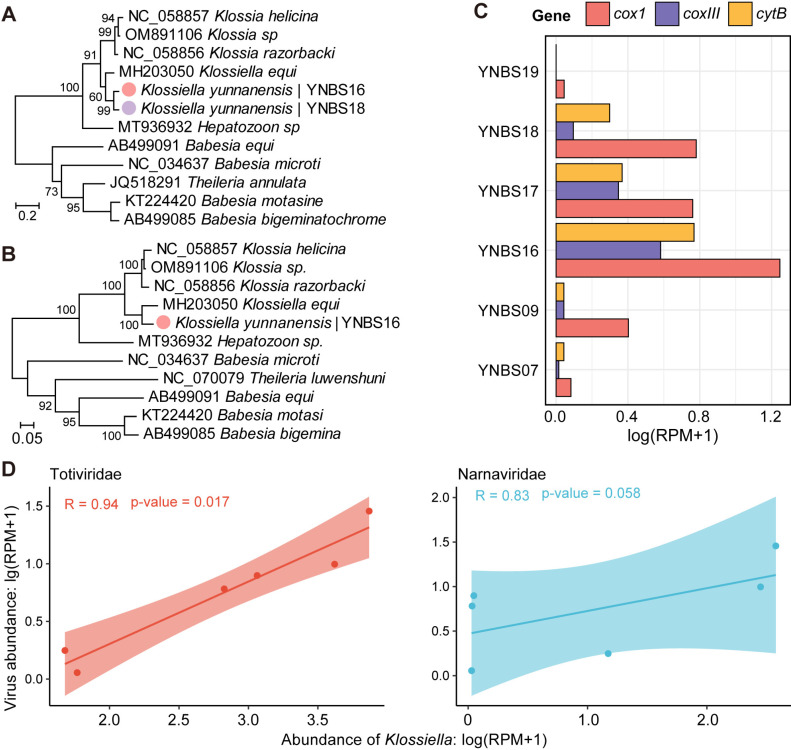
Bats are natural reservoirs for a wide range of microorganisms, including many notable zoonotic pathogens. However, the composition of the infectome (i.e., the collection of viral, bacterial and eukaryotic microorganisms) within bat kidneys remains poorly understood. To address this gap, we performed meta-transcriptomic sequencing on kidney tissues from 142 bats, spanning ten species sampled at five locations in Yunnan province, China. This analysis identified 22 viral species, including 20 novel viruses, two of which represented newly discovered henipaviruses closely related to the highly pathogenic Hendra and Nipah viruses. These henipaviruses were found in the kidneys of bats inhabiting an orchard near villages, raising concerns about potential fruit contamination via bat urine and transmission risks to livestock or humans. Additionally, we identified a novel protozoan parasite, tentatively named Klossiella yunnanensis, along with two highly abundant bacterial species, one of which is a newly discovered species-Flavobacterium yunnanensis. These findings broaden our understanding of the bat kidney infectome, underscore critical zoonotic threats, and highlight the need for comprehensive, full-spectrum microbial analyses of previously understudied organs to better assess spillover risks from bat populations.
Copyright: © 2025 Kuang et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
From Bat to Worse: The Pivotal Role of Bats for Viral Zoonosis.Microb Biotechnol. 2025 Jul;18(7):e70190. doi: 10.1111/1751-7915.70190. Microb Biotechnol. 2025. PMID: 40619741 Free PMC article. Review.
-
A Comparative Assessment of the Pathogenic Potential of Newly Discovered Henipaviruses.Pathogens. 2024 Jul 16;13(7):587. doi: 10.3390/pathogens13070587. Pathogens. 2024. PMID: 39057814 Free PMC article. Review.
-
Domesticated animals as hosts of henipaviruses and filoviruses: A systematic review.Vet J. 2018 Mar;233:25-34. doi: 10.1016/j.tvjl.2017.12.024. Epub 2017 Dec 30. Vet J. 2018. PMID: 29486875
-
Discovery and Genomic Characterization of a Novel Henipavirus, Angavokely Virus, from Fruit Bats in Madagascar.J Virol. 2022 Sep 28;96(18):e0092122. doi: 10.1128/jvi.00921-22. Epub 2022 Aug 30. J Virol. 2022. PMID: 36040175 Free PMC article.
-
Laboratory Diagnosis of Hendra and Nipah: Two Emerging Zoonotic Diseases with One Health Significance.Viruses. 2025 Jul 17;17(7):1003. doi: 10.3390/v17071003. Viruses. 2025. PMID: 40733619 Free PMC article. Review.
References
-
- Simmons NB, Cirranello AL. Bat Species of the World: A taxonomic and geographic database. Version 1.6. 2024. https://zenodo.org/records/12802826
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
