

Bifidobacterium deficit in United States infants drives prevalent gut dysbiosis
- PMID: 40555747
- PMCID: PMC12187928
- DOI: 10.1038/s42003-025-08274-7
Bifidobacterium deficit in United States infants drives prevalent gut dysbiosis
Abstract
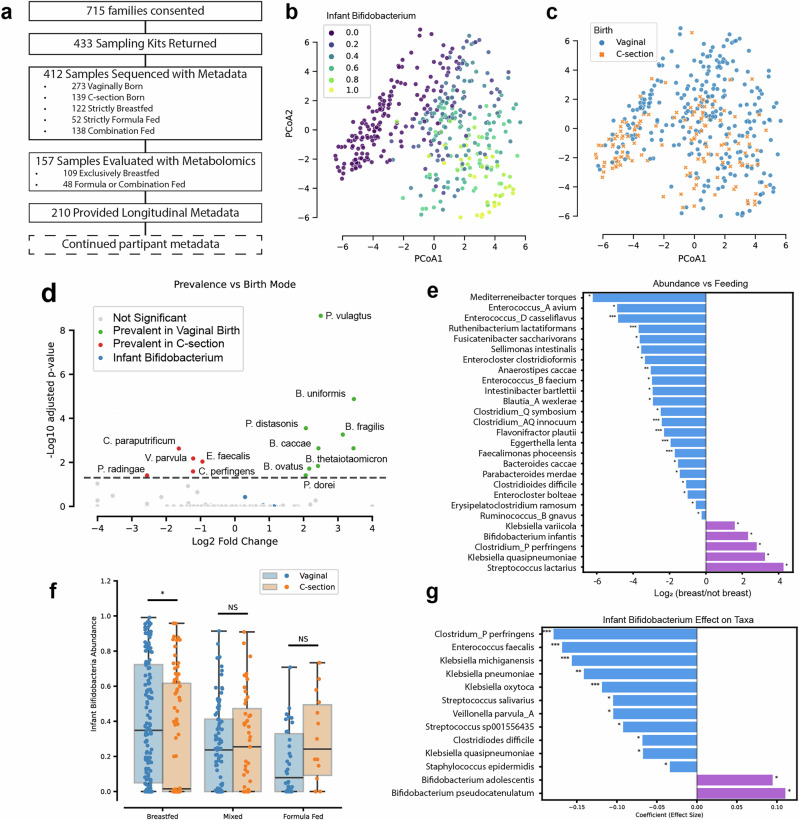
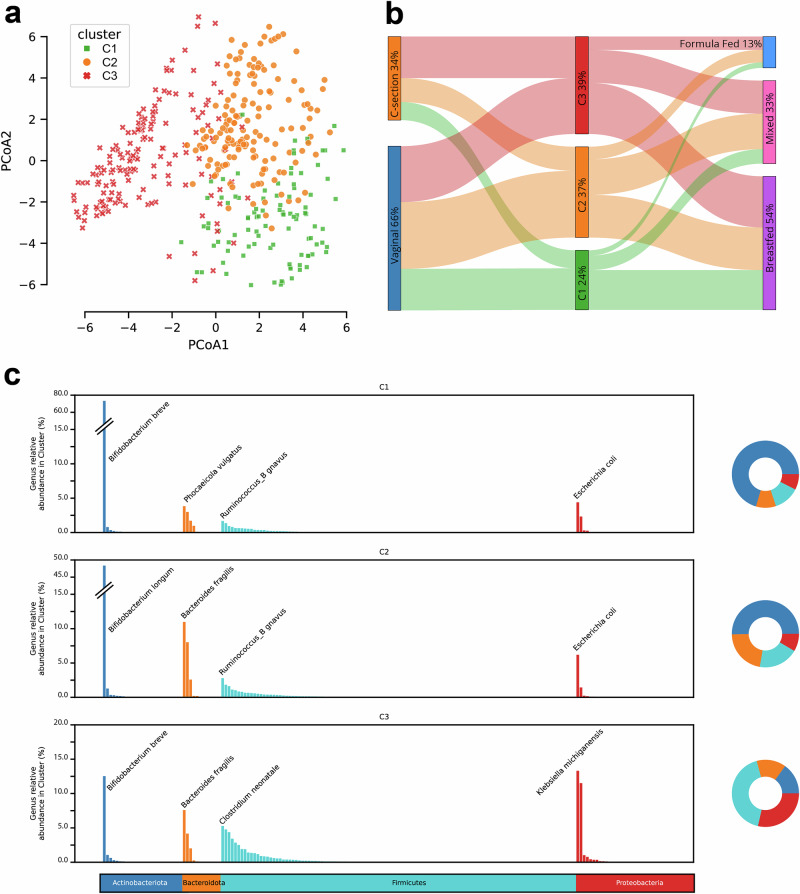
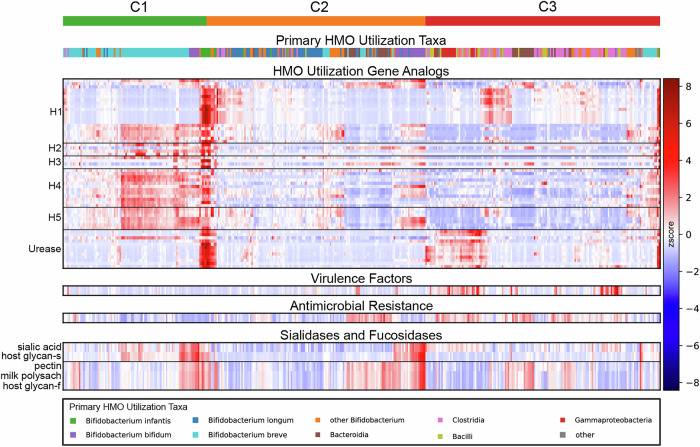
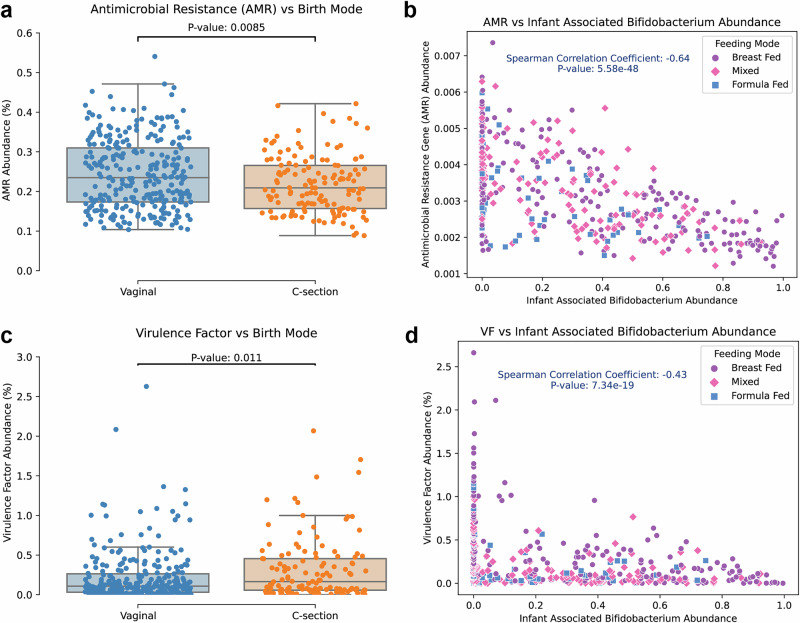
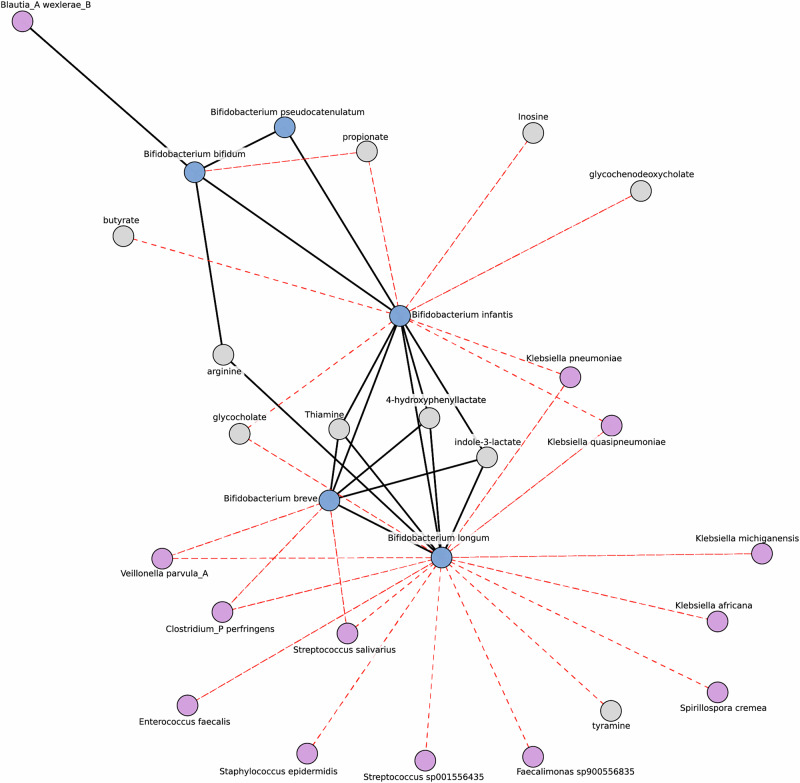
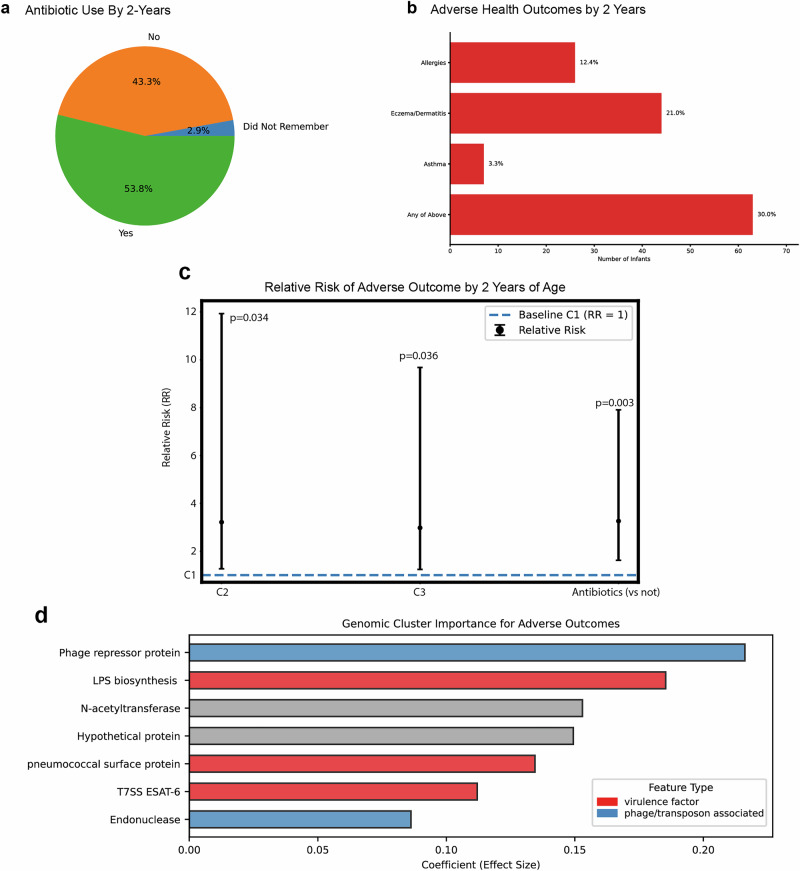
The composition of the infant gut microbiome is critical to immune development and noncommunicable disease (NCD) trajectory. However, a comprehensive evaluation of the infant gut microbiome in the United States is lacking. The My Baby Biome study, designed to address this knowledge gap, evaluated the gut microbiomes of 412 infants (representative of U.S. demographic diversity) using metagenomics and metabolomics. Regardless of birth mode and/or feeding method, widespread Bifidobacterium deficit was observed, with approximately 25% of U.S. infants lacking detectable Bifidobacterium. Bifidobacterium-dominant microbiomes exhibit distinct features when compared to microbiomes with other dominant microbial compositions including reduced antimicrobial resistance and virulence factor genes, altered carbohydrate utilization pathways, and altered metabolic signatures. In C-section birth infants, Bifidobacterium tended to be replaced in the human milk oligosaccharide utilization niche with potentially pathogenic species. Longitudinal health outcomes from these infants suggest that the disappearance of key Bifidobacterium may contribute to the development of atopy.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: Research was funded by Persephone Biosciences. All authors were employed by and/or hold stock in Persephone Biosciences. RI also holds stock in Kenvue.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
