

Optimizing genomic diversity assessments for conservation of Bromus auleticus (Trinius ex Nees) using individual and pooled sequencing
- PMID: 40560985
- PMCID: PMC12194279
- DOI: 10.1371/journal.pone.0325548
Optimizing genomic diversity assessments for conservation of Bromus auleticus (Trinius ex Nees) using individual and pooled sequencing
Abstract
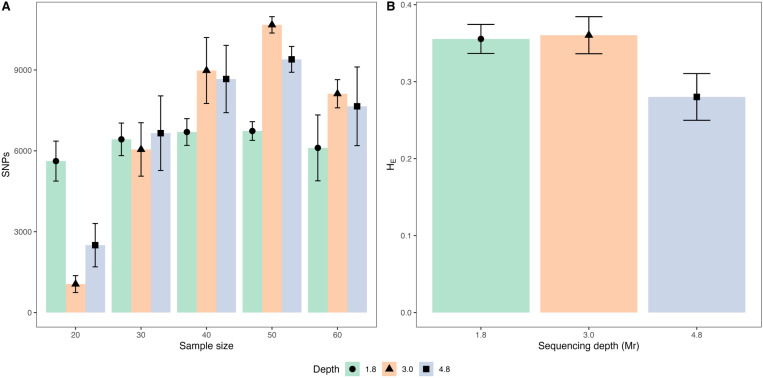
Bromus auleticus, a valuable forage grass native to the Pampa biome, is currently undergoing genetic erosion. Therefore, it is essential to assess appropriate methodologies for developing population genomic studies that will contribute to the conservation of this genetic resource. In this study, we evaluated five accessions using two genotyping strategies: individual sequencing (ind-seq) and pooled sequencing (pool-seq). To assess methodologies effectiveness, the correlation between allele frequencies calculated using each approach was investigated, as well as genetic diversity and population structure. These comparisons explicitly accounted for the potential effects of factors such as sample size, missing data, sequencing depth, and minor allele frequencies. The highest values of frequencies concordance and percentage of SNPs in common between ind-seq and pool-seq were achieved using a sample size of 30-60 plants per accession. These values were obtained with a maximum missing data threshold of 10% and a less strict minimum allele frequency threshold for pool-seq (0.01) compared to ind-seq (0.05). Pool-seq required a higher sequencing depth per accession (4.8 million reads) compared to ind-seq (0.9 million reads) to achieve similar allele frequencies. Pools of 50 individuals yielded the highest number of polymorphic sites, averaging over 9,000 per accession at a sequencing depth of 4.8 Mr. Under these conditions, pool-seq consistently resulted in an average of 0.09 higher expected heterozygosity and a 0.24 lower allelic richness compared to ind-seq in all accessions. Population structure inferred with both methodologies confirmed the outcrossing nature of B. auleticus and aligned with the geographical origin of each accession. The average inbreeding coefficient of 0.2 evidence inbreeding, which highlights the importance of conservation efforts for this valuable plant genetic resource. Based on these findings, we propose two workflows for conducting population genomics studies on Bromus auleticus.
Copyright: © 2025 Gillman et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures









References
-
- Bilenca D, Miñarro F. Identificación de áreas valiosas de pastizal (AVPs) en las pampas y campos de Argentina [Identification of valuable grassland areas (VPA) in the pampas and campos of Argentina]. Fundación Vida Silvestre Argentina, ed. Buenos Aires: Fundación Vida Silvestre Argentina; 2004. p. 353.
-
- Olmos F. Bromus auleticus. First ed. Unidad de difusión e información tecnológica del INIA, ed. Montevideo: Unidad de difusión e información tecnológica del INIA; 1993. p. 30
-
- Global core biodata resource. 2024. https://www.gbif.org/es/species/4107276. - PMC - PubMed
-
- Williams WM, Stewart AV, Williamson ML. Bromus. In: Kole C, ed. Wild crop relatives: genomic and breeding resources, millets and grasses. Berlin, Heidelberg: Springer-Verlag Berlin Heidelberg; 2011. p. 15–30.
MeSH terms
LinkOut - more resources
Full Text Sources
