

Mapping MAVE data for use in human genomics applications
- PMID: 40563119
- PMCID: PMC12188674
- DOI: 10.1186/s13059-025-03647-x
Mapping MAVE data for use in human genomics applications
Abstract
Background: Experimental data from functional assays have a critical role in interpreting the impact of genetic variants. Assay data must be unambiguously mapped to a reference genome to make it accessible, but it is often reported relative to assay-specific sequences, complicating downstream use and integration of variant data across resources. To make multiplexed assays of variant effect (MAVE) data more broadly available to the research and clinical communities, the Atlas of Variant Effects Alliance mapped MAVE data from the MaveDB community database to human reference sequences, creating an extensive set of machine-readable homology mappings that are incorporated into widely used human genomics applications.
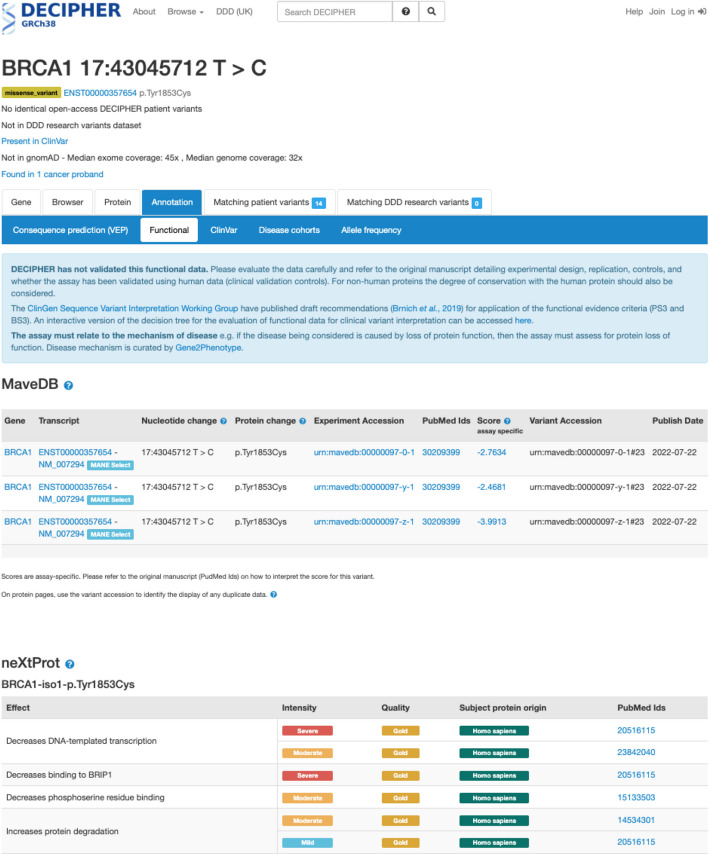
Results: Here, we map approximately 9.0 million individual protein and nucleotide variants in MaveDB to the human genome, describing the examined variants with respect to human reference sequences while preserving the data provenance of the original MAVE sequences. We then disseminate the results to major genomic resources including the Genomics 2 Proteins Portal, UCSC Genome Browser, Ensembl Variant Effect Predictor, and DECIPHER platform. Within these applications, MAVE variants can now be visualized and integrated with other relevant clinical and biological data, making additional knowledge available when performing variant interpretation and conducting other research activities.
Conclusions: Mapping MAVE variants to human reference sequences and sharing the mapped dataset with several key human genomics applications enables a new and diverse set of applications for MAVE data. This study provides increased access to functional data that can assist in clinical variant interpretation pipelines and enable biomedical research and discovery.
Keywords: Deep mutational scanning; Functional assay; Genomic medicine; Genomics; Global Alliance for Genomics and Health; Massively parallel reporter assays; Multiplexed assays of variant effect; Variation representation specification.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare that they have no competing interests. Review history: The review history is available as Additional file 7. Peer review information: Yang Li and Veronique van den Berghe were the primary editors of this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Figures




Update of
-
Mapping MAVE data for use in human genomics applications.bioRxiv [Preprint]. 2024 Jun 30:2023.06.20.545702. doi: 10.1101/2023.06.20.545702. bioRxiv. 2024. Update in: Genome Biol. 2025 Jun 25;26(1):179. doi: 10.1186/s13059-025-03647-x. PMID: 38979347 Free PMC article. Updated. Preprint.
References
-
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. - PMC - PubMed
MeSH terms
Grants and funding
- U01CA242954/CA/NCI NIH HHS/United States
- R35 HG011949/HG/NHGRI NIH HHS/United States
- RM1 HG010461/HG/NHGRI NIH HHS/United States
- UM1HG011969/HG/NHGRI NIH HHS/United States
- U01 CA242954/CA/NCI NIH HHS/United States
- U41 HG009649/HG/NHGRI NIH HHS/United States
- NIHR203312/NIHR Cambridge Biomedical Research Centre
- U24HG002371/HG/NHGRI NIH HHS/United States
- U24 HG009649/HG/NHGRI NIH HHS/United States
- HG009649/HG/NHGRI NIH HHS/United States
- UM1 HG011969/HG/NHGRI NIH HHS/United States
- U24 HG002371/HG/NHGRI NIH HHS/United States
- RM1HG010461/HG/NHGRI NIH HHS/United States
- R35HG011949/HG/NHGRI NIH HHS/United States
