

This is a preprint.
Path2Omics: Enhanced transcriptomic and methylation prediction accuracy from tumor histopathology
- PMID: 40568160
- PMCID: PMC12190742
- DOI: 10.1101/2025.02.26.640189
Path2Omics: Enhanced transcriptomic and methylation prediction accuracy from tumor histopathology
Update in
-
Path2Omics Enhances Transcriptomic and Methylation Prediction Accuracy from Tumor Histopathology.Cancer Res. 2025 Oct 30. doi: 10.1158/0008-5472.CAN-25-4350. Online ahead of print. Cancer Res. 2025. PMID: 41166699
Abstract
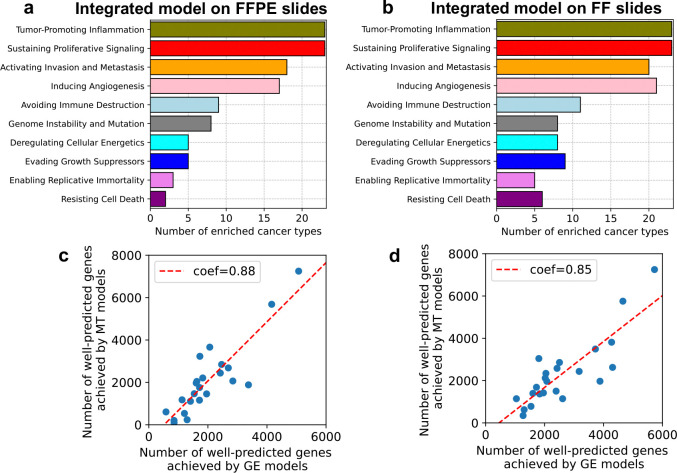
Precision oncology is becoming increasingly integral to clinical practice, demonstrating notable improvements in treatment outcomes. While molecular data provide comprehensive insights, obtaining such data remains costly and time-consuming. To address this challenge, we developed Path2Omics, a deep learning model that predicts gene expression and methylation from histopathology for 23 cancer types. Path2Omics was trained on 20,497 slides (9,456 formalin-fixed and paraffin-embedded (FFPE) and 11,041 fresh frozen (FF)) from 8,007 patients across 23 The Cancer Genome Atlas cohorts. When tested on FFPE slides, the most readily available format in clinical pathology practice, the integrated model outperformed its individual FF and FFPE components, robustly predicting nearly 5,000 genes on average, approximately five times more than our recently published DeepPT model. Externally evaluated on seven independent cohorts, Path2Omics robustly predicted the expression of approximately 4,400 genes, yielding a 30% increase over the FFPE model alone. Finally, we demonstrate that the inferred gene expression is nearly as effective as the actual values in predicting patient survival and treatment response. These results lay the basis for using Path2Omics to advance precision oncology from histopathology slides in a speedy and cost-effective manner.
Conflict of interest statement
Competing interest E.R. is a co-founder of Medaware, Metabomed and Pangea Biomed (divested from the latter). E.R. serves as a non-paid scientific consultant to Pangea Biomed under a collaboration agreement between Pangea Biomed and the NCI. E.R. also serves as a scientific advisory board member of GSK oncology. The other authors declare no competing interests.
Figures













References
-
- Beck Andrew H., Sangoi Ankur R., Leung Samuel, Marinelli Robert J., Nielsen Torsten O., van de Vijver Marc J., West Robert B., van de Rijn Matt, and Koller Daphne. 2011. “Systematic Analysis of Breast Cancer Morphology Uncovers Stromal Features Associated with Survival.” Science Translational Medicine 3 (108): 108ra113. - PubMed
-
- Boehm Kevin M., Aherne Emily A., Ellenson Lora, Nikolovski Ines, Alghamdi Mohammed, Vázquez-García Ignacio, Zamarin Dmitriy, et al. 2022. “Multimodal Data Integration Using Machine Learning Improves Risk Stratification of High-Grade Serous Ovarian Cancer.” Nature Cancer 3 (6): 723–33. - PMC - PubMed
-
- Bulten Wouter, Pinckaers Hans, van Boven Hester, Vink Robert, de Bel Thomas, van Ginneken Bram, van der Laak Jeroen, Hulsbergen-van de Kaa Christina, and Litjens Geert. 2020. “Automated Deep-Learning System for Gleason Grading of Prostate Cancer Using Biopsies: A Diagnostic Study.” The Lancet Oncology 21 (2): 233–41. - PubMed
Publication types
LinkOut - more resources
Full Text Sources