

Gastrointestinal inflammation and cancer: viral and bacterial interplay
- PMID: 40568785
- PMCID: PMC12203856
- DOI: 10.1080/19490976.2025.2519703
Gastrointestinal inflammation and cancer: viral and bacterial interplay
Abstract
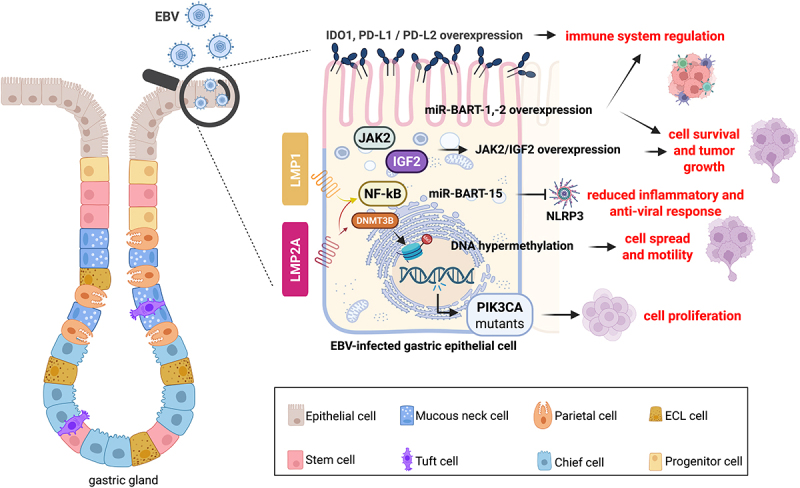
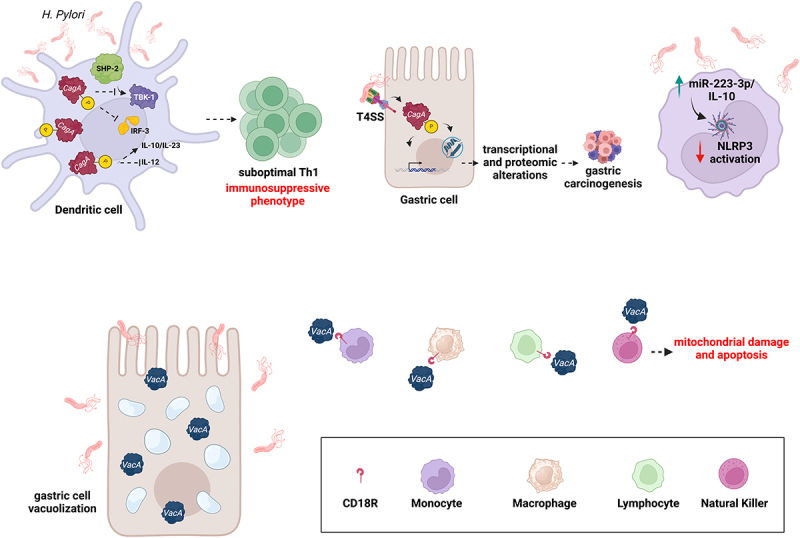
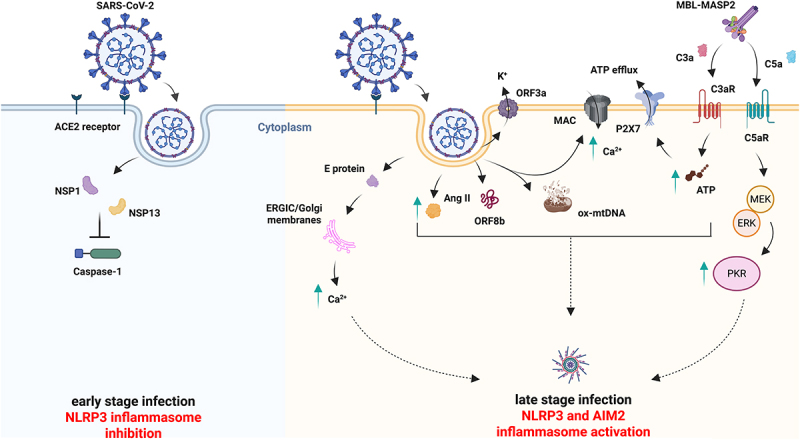
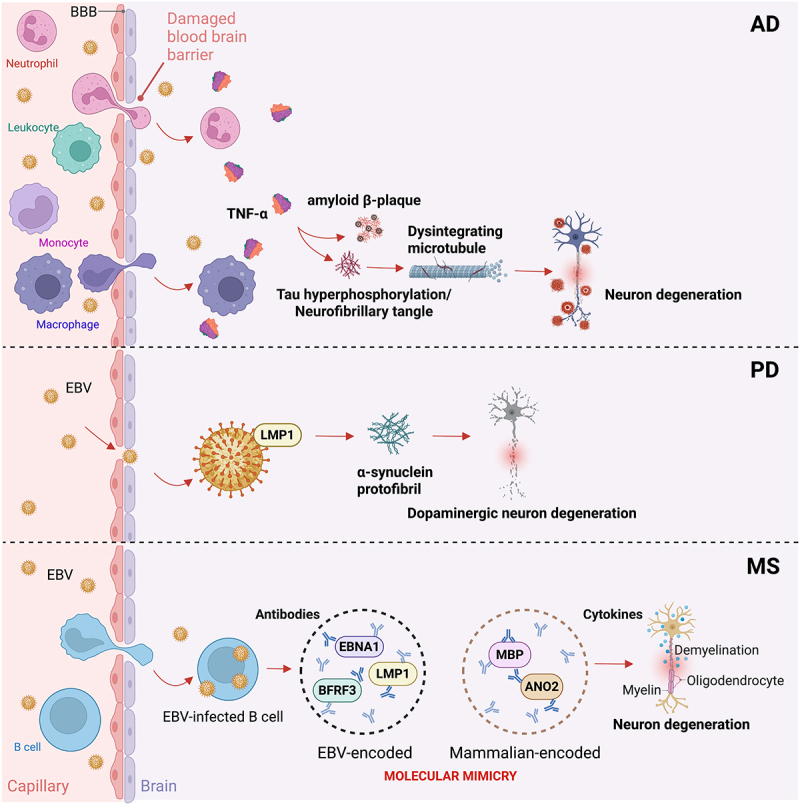
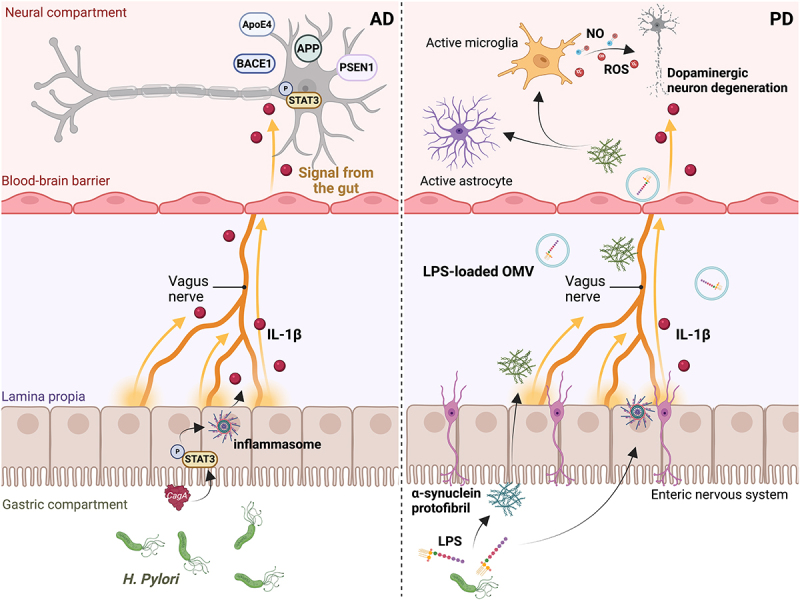
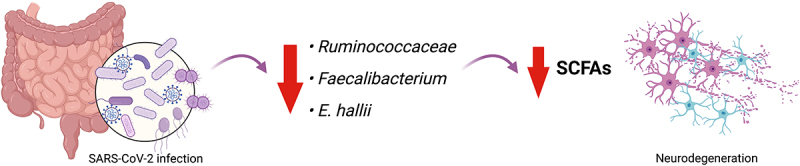
Gastrointestinal (GI) inflammation and malignancies arise from complex interactions between the host's immune responses and microbial pathogens. Epstein-Barr virus (EBV), Helicobacter pylori (H. pylori), and Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) contribute to chronic GI inflammation, immune evasion, and tumorigenesis through distinct but interconnected mechanisms. EBV, a widespread herpesvirus, establishes a latent infection in B cells and epithelial cells. It promotes gastric carcinogenesis through immune modulation, epigenetic changes, and viral microRNAs (miRNAs). H. pylori, a gastric carcinogen, induces chronic gastritis and gastric cancer (GC) through Cytotoxin-associated gene A (CagA) and Vacuolating cytotoxin gene A (VacA) virulence factors. These factors disrupt host immune responses and enhance oncogenic signaling pathways. Recent evidence also links SARS-CoV-2 to gut dysbiosis and inflammatory responses. It worsens immune dysfunction and hence potentially impacting GI pathology. EBV and H. pylori co-infections may synergistically amplify inflammatory signaling, creating a tumor-promoting microenvironment. This review emphasizes the molecular mechanisms by which these pathogens contribute to GI diseases, focusing on their immune evasion strategies and potential therapeutic targets. Understanding these interactions is essential for developing targeted interventions for infection-driven GI malignancies.
Keywords: EBV; H. Pylori; NLRP3; SARS-CoV-2; cancer; gastrointestinal disease; immune escape; inflammation; microRNA.
Conflict of interest statement
The authors declare the absence of any commercial or financial conflict of interest.
Figures
Similar articles
-
Advances in Gastric Carcinogenesis Related to Helicobacter Pylori.Chirurgia (Bucur). 2025 Jun;120(3):322-344. doi: 10.21614/chirurgia.3147. Chirurgia (Bucur). 2025. PMID: 40637070 Review.
-
Impact of helicobacter pylori infection on the efficacy of ICIs in gastric cancer.BMC Cancer. 2025 Jul 1;25(1):1101. doi: 10.1186/s12885-025-14436-x. BMC Cancer. 2025. PMID: 40597779 Free PMC article. Review.
-
Helicobacter pylori and Epstein-Barr Virus Coinfection Stimulates Aggressiveness in Gastric Cancer through the Regulation of Gankyrin.mSphere. 2021 Oct 27;6(5):e0075121. doi: 10.1128/mSphere.00751-21. Epub 2021 Sep 29. mSphere. 2021. PMID: 34585958 Free PMC article.
-
Effect of Helicobacter Pylori infection on immunotherapy for gastrointestinal cancer: a narrative review.Immunotherapy. 2025 Apr;17(5):355-368. doi: 10.1080/1750743X.2025.2479410. Epub 2025 Mar 14. Immunotherapy. 2025. PMID: 40087147 Review.
-
Epstein Barr virus and Helicobacter pylori co-infection are positively associated with severe gastritis in pediatric patients.PLoS One. 2013 Apr 24;8(4):e62850. doi: 10.1371/journal.pone.0062850. Print 2013. PLoS One. 2013. PMID: 23638154 Free PMC article.
References
-
- Burd EM, Hinrichs BH.. Gastrointestinal infections. Mol Pathol Clin Pract. 2015; 707–30. doi: 10.1007/978-3-319-19674-9_50. - DOI
-
- Ayoubian H, Ludwig N, Fehlmann T, Menegatti J, Gröger L, Anastasiadou E, Trivedi P, Keller A, Meese E, Grässer FA, et al. Epstein-Barr virus infection of cell lines derived from diffuse large B-Cell lymphomas alters MicroRNA loading of the Ago2 complex. J Virol. 2019. Jan 17. 93(3):e01297–18. doi: 10.1128/JVI.01297-18. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous