

TAS-seq enables subcellular single-stranded adenosine profiling by signal peptide-assisted adenosine deamination
- PMID: 40570838
- PMCID: PMC12296453
- DOI: 10.1016/j.crmeth.2025.101087
TAS-seq enables subcellular single-stranded adenosine profiling by signal peptide-assisted adenosine deamination
Abstract
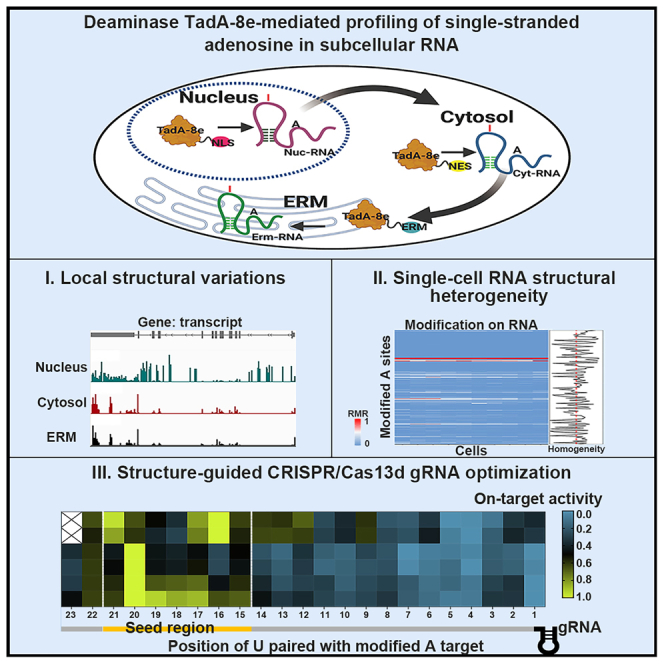
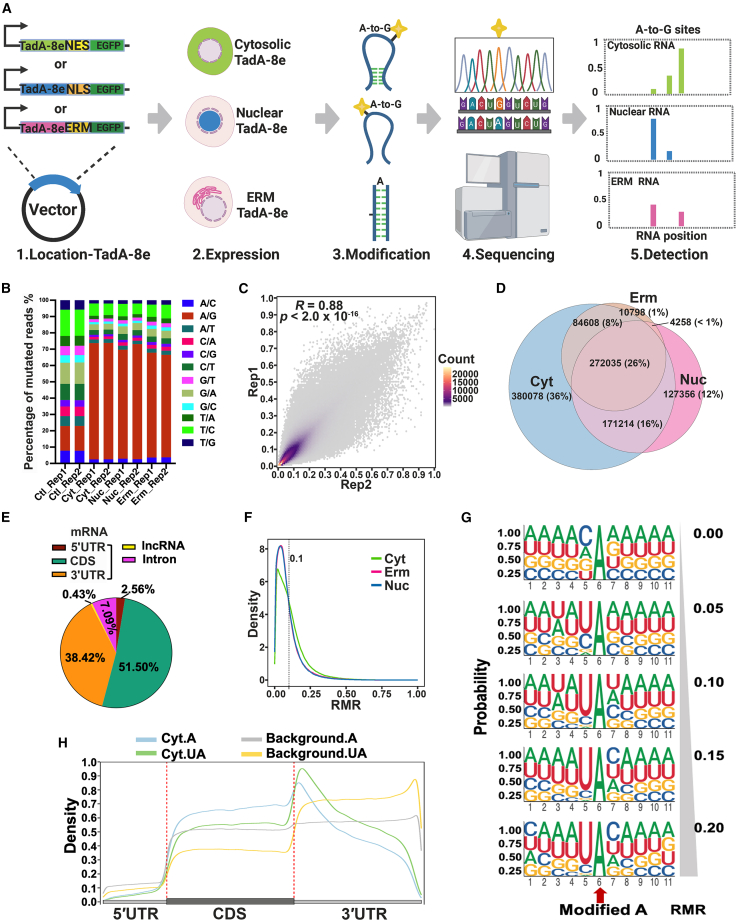
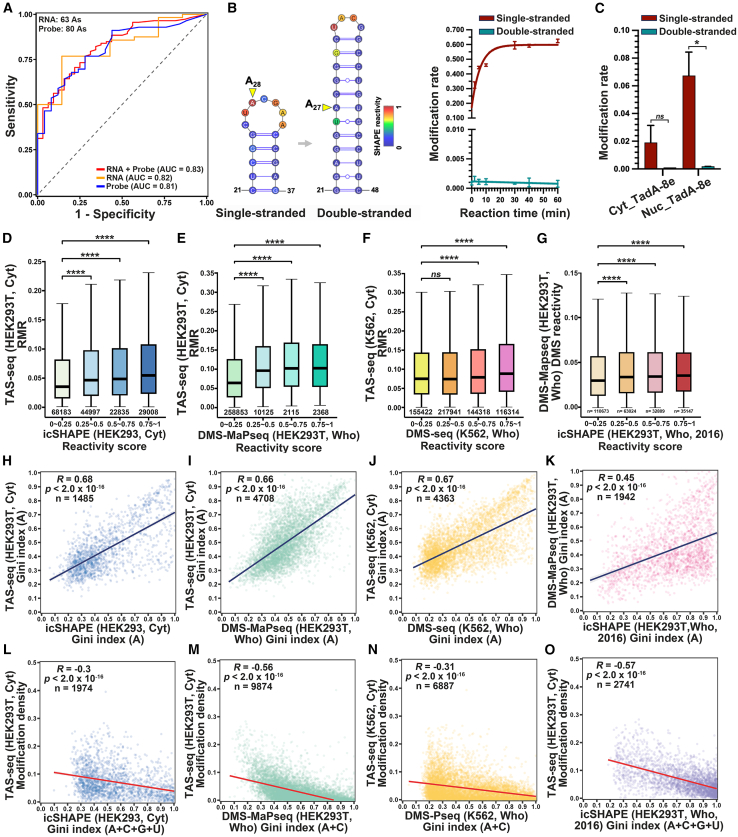
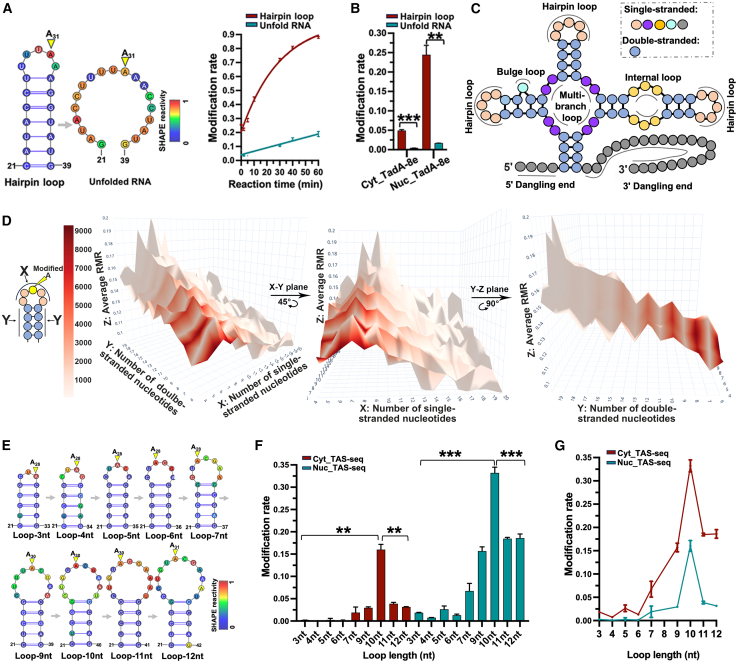
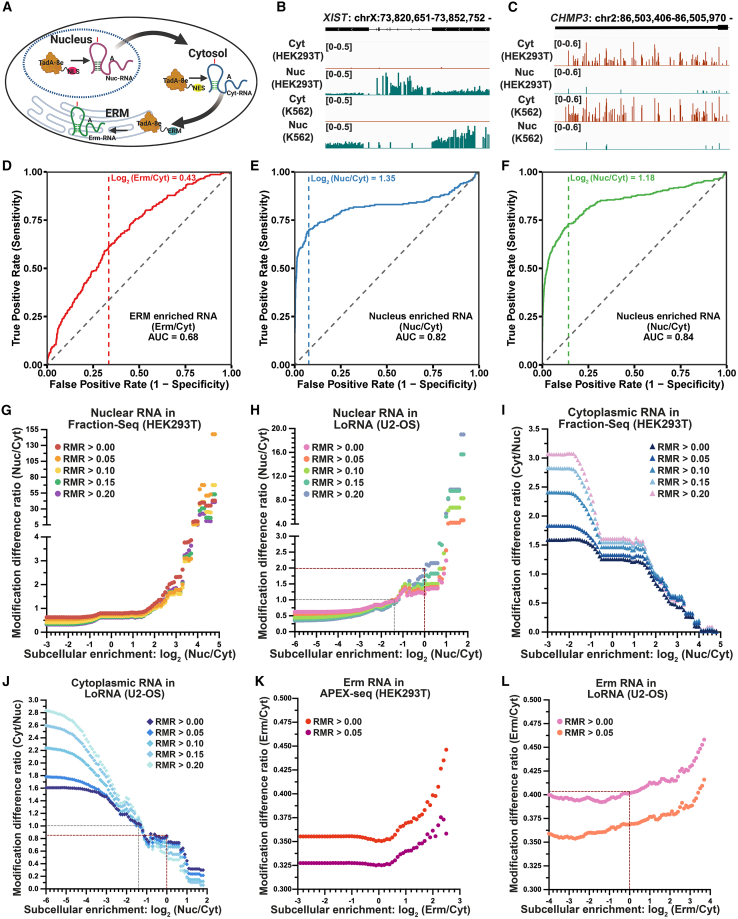
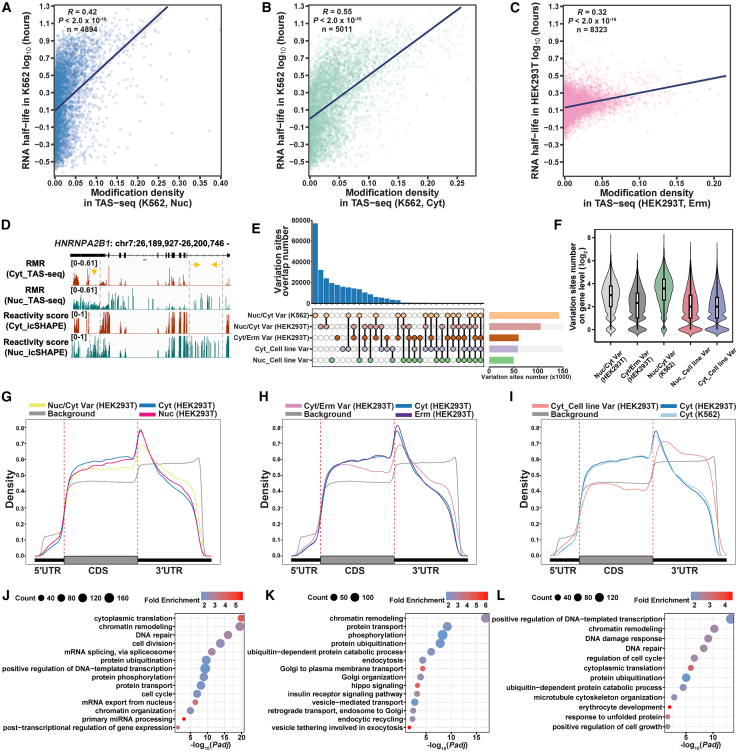
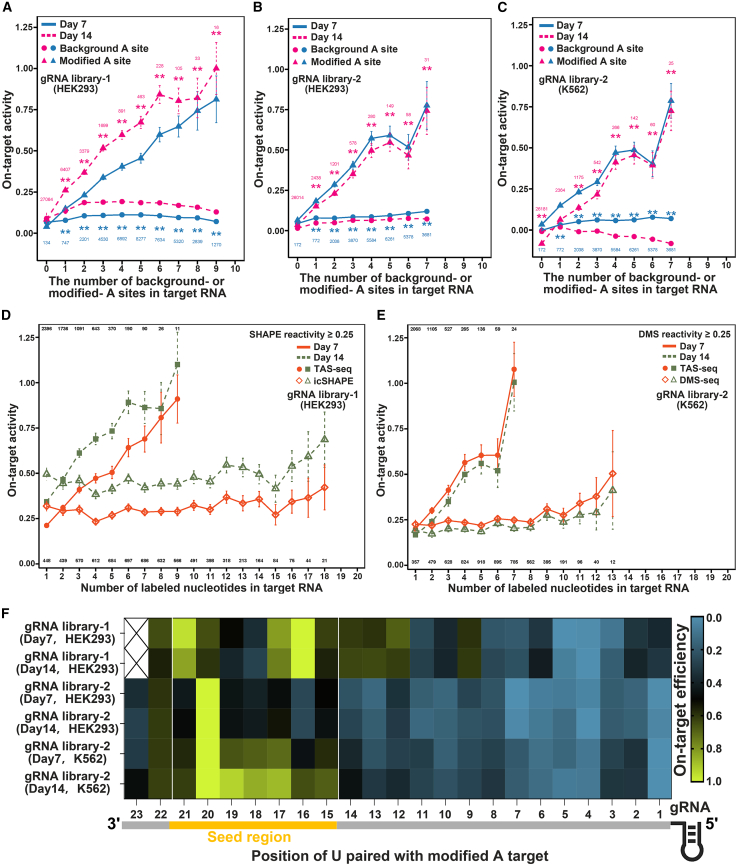
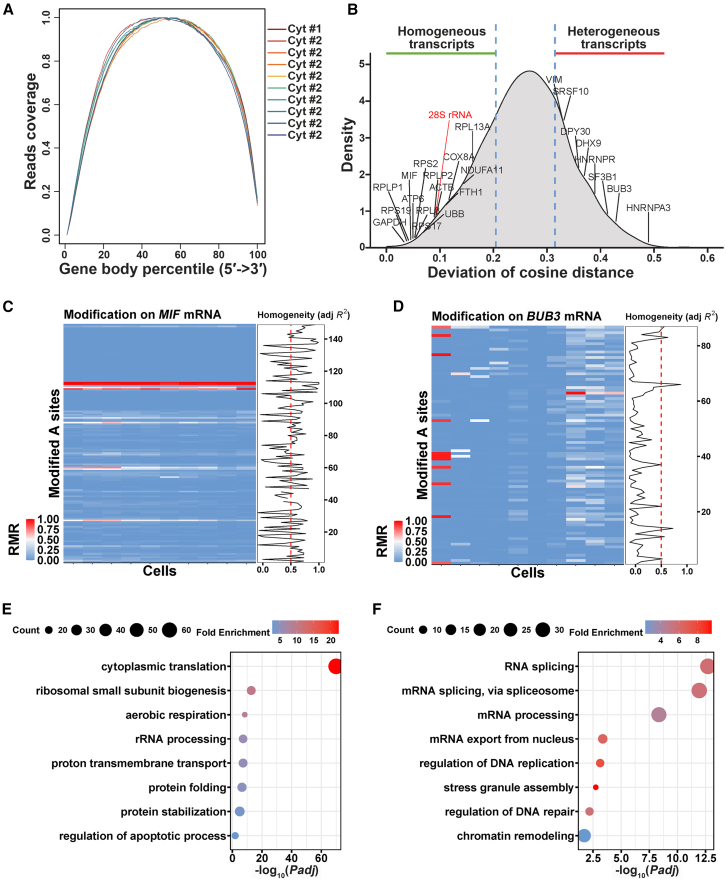
RNA structure plays a crucial role in its function and undergoes dynamic changes throughout its life cycle. To study these dynamics, we developed TAS sequencing (TAS-seq), which expresses the deaminase TadA-8e in specific subcellular compartments to modify single-stranded adenosines, particularly within hairpin loops. We applied TAS-seq to the nucleus, cytosol, and endoplasmic reticulum membrane, identifying adenosine structural variations and compartment-specific regulation of RNA stability. Single-cell TAS-seq revealed structural heterogeneity of cytosolic RNAs. Additionally, adenosines labeled by TAS-seq contribute to guide RNA optimization in the CRISPR-Cas13d system. Our method provides insights into compartment-specific RNA structural dynamics, cell-specific heterogeneity, and their functional implications.
Keywords: CP: cell biology; CP: molecular biology; RNA structural dynamics; gRNA structure and performance; subcellular adenosine structure profiling.
Copyright © 2025 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Similar articles
-
Multimodal CRISPR screens uncover DDX39B as a global repressor of A-to-I RNA editing.Cell Rep. 2025 Jul 22;44(7):116009. doi: 10.1016/j.celrep.2025.116009. Epub 2025 Jul 12. Cell Rep. 2025. PMID: 40652511
-
Soft graph clustering for single-cell RNA sequencing data.BMC Bioinformatics. 2025 Jul 25;26(1):195. doi: 10.1186/s12859-025-06231-z. BMC Bioinformatics. 2025. PMID: 40713495 Free PMC article.
-
A scRNA-seq reference contrasting living and early post-mortem human retina across diverse donor states.Hum Genomics. 2025 Jul 14;19(1):81. doi: 10.1186/s40246-025-00796-9. Hum Genomics. 2025. PMID: 40660409 Free PMC article.
-
Single-cell RNA sequencing (scRNA-seq) and its insights into cellular heterogeneity in atherosclerosis.Vascul Pharmacol. 2025 Jun;159:107499. doi: 10.1016/j.vph.2025.107499. Epub 2025 May 7. Vascul Pharmacol. 2025. PMID: 40345606 Review.
-
Unraveling asthma through single-cell RNA sequencing in understanding disease mechanisms.J Asthma. 2025 Aug;62(8):1257-1265. doi: 10.1080/02770903.2025.2472358. Epub 2025 Mar 7. J Asthma. 2025. PMID: 40014380 Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
