

Characterisation and prevalence of inherited retinal diseases in the Finnish population reveals enrichment of population-specific phenotypes and causative variants
- PMID: 40571344
- PMCID: PMC12320592
- DOI: 10.1136/bjo-2025-327427
Characterisation and prevalence of inherited retinal diseases in the Finnish population reveals enrichment of population-specific phenotypes and causative variants
Abstract
Aims: This study aims to assess clinical and genetic characteristics as well as the prevalence of inherited retinal dystrophies (IRD) and their subphenotypes in the Finnish founder population.
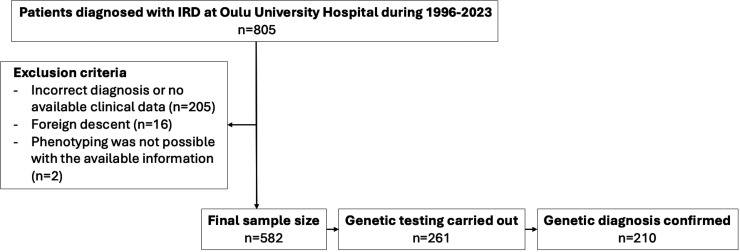
Methods: A retrospective analysis of clinical and genetic data from Northern Finnish patients diagnosed with IRD between 1996 and 2023 at Oulu University Hospital, Finland, was conducted.
Results: The cohort comprised 582 patients with IRD, categorised into 16 different subphenotypes. Pathogenic or likely pathogenic variants explaining IRD were identified in 36% (n=210/582) of all patients and 80% (n=210/261) of genetically tested patients with IRD. Diagnostic yields varied between different IRD subphenotypes. The genetic aetiology was most commonly confirmed in X-linked retinoschisis, severe early childhood-onset retinal dystrophy, congenital stationary night blindness and choroideremia. The lowest rates of causative variant identification were observed in cone or cone-rod dystrophy and macular dystrophy. In total, 70 pathogenic or likely pathogenic variants were identified across 39 different genes; variants in the FZD4 and RPGR genes were the most prevalent. Over half of the variants were enriched in the Finnish population. The estimated total prevalence of IRDs in Northern Finland was 69.8/100 000 (1:1432). The prevalence of the most common subphenotypes was as follows: retinitis pigmentosa, 25.3/100 000; X-linked retinoschisis, 10.7/100 000; Usher syndrome, 8.9/100 000; choroideremia, 7/100 000 and cone or cone-rod dystrophy, 6/100 000.
Conclusion: The Northern Finnish population exhibits an enrichment of population-specific IRD-associated variants, resulting in a high overall prevalence of IRDs and an increased prevalence of selected retinal subphenotypes, such as retinoschisis, choroideremia and Usher syndrome types 3 and 1.
Keywords: Diagnostic tests/Investigation; Dystrophy; Epidemiology; Genetics; Retina.
© Author(s) (or their employer(s)) 2025. Re-use permitted under CC BY. Published by BMJ Group.
Conflict of interest statement
Competing interests: None declared.
Figures


Similar articles
-
Genetic landscape of inherited retinal dystrophies in a Welsh tertiary referral centre.Br J Ophthalmol. 2025 Jul 22;109(8):845-851. doi: 10.1136/bjo-2024-327049. Br J Ophthalmol. 2025. PMID: 40541286
-
Exploring Concomitant Ophthalmic Comorbidities in Portuguese Patients with Inherited Retinal Diseases: A Comprehensive Clinical Study.Genes (Basel). 2025 Jun 26;16(7):743. doi: 10.3390/genes16070743. Genes (Basel). 2025. PMID: 40725401 Free PMC article.
-
Clinical exome analysis and targeted gene repair of the c.1354dupT variant in iPSC lines from patients with PROM1-related retinopathies exhibiting diverse phenotypes.Stem Cell Res Ther. 2024 Jul 2;15(1):192. doi: 10.1186/s13287-024-03804-2. Stem Cell Res Ther. 2024. PMID: 38956727 Free PMC article.
-
Epidemiology of Mutations in the 65-kDa Retinal Pigment Epithelium (RPE65) Gene-Mediated Inherited Retinal Dystrophies: A Systematic Literature Review.Adv Ther. 2022 Mar;39(3):1179-1198. doi: 10.1007/s12325-021-02036-7. Epub 2022 Jan 30. Adv Ther. 2022. PMID: 35098484 Free PMC article.
-
The Diagnostic Yield of Next Generation Sequencing in Inherited Retinal Diseases: A Systematic Review and Meta-analysis.Am J Ophthalmol. 2023 May;249:57-73. doi: 10.1016/j.ajo.2022.12.027. Epub 2022 Dec 30. Am J Ophthalmol. 2023. PMID: 36592879
References
-
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources