

Compensatory evolution to DNA replication stress is robust to nutrient availability
- PMID: 40571846
- PMCID: PMC12494895
- DOI: 10.1038/s44320-025-00127-z
Compensatory evolution to DNA replication stress is robust to nutrient availability
Abstract
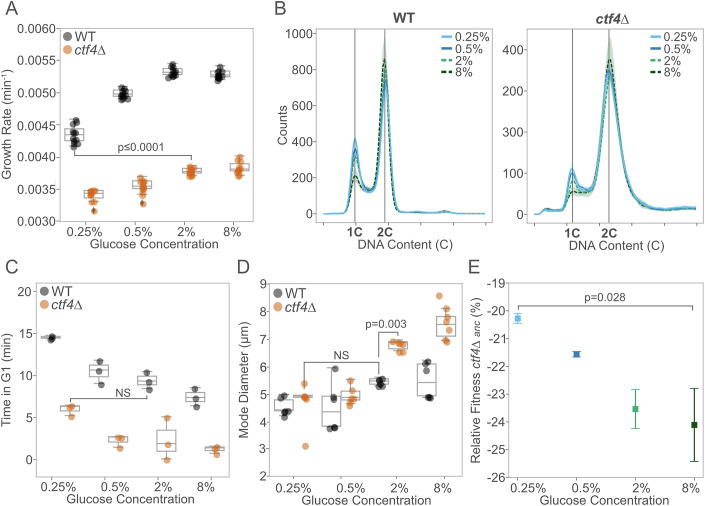
Evolutionary repair refers to the compensatory evolution that follows perturbations in cellular processes. While evolutionary trajectories are often reproducible, other studies suggest they are shaped by genotype-by-environment (GxE) interactions. Here, we test the predictability of evolutionary repair in response to DNA replication stress-a severe perturbation impairing the conserved mechanisms of DNA synthesis, resulting in genetic instability. We conducted high-throughput experimental evolution on Saccharomyces cerevisiae experiencing constitutive replication stress, grown under different glucose availability. We found that glucose levels impact the physiology and adaptation rate of replication stress mutants. However, the genetics of adaptation show remarkable robustness across environments. Recurrent mutations collectively recapitulated the fitness of evolved lines and are advantageous across macronutrient availability. We also identified a novel role of the mediator complex of RNA polymerase II in adaptation to replicative stress. Our results highlight the robustness and predictability of evolutionary repair mechanisms to DNA replication stress and provide new insights into the evolutionary aspects of genome stability, with potential implications for understanding cancer development.
Keywords: S. cerevisiae; Compensatory Evolution; DNA Replication Stress; Genome Maintenance; Nutrients.
© 2025. The Author(s).
Conflict of interest statement
Disclosure and competing interests statement. The authors declare no competing interests.
Figures










Update of
-
Compensatory Evolution to DNA Replication Stress is Robust to Nutrient Availability.bioRxiv [Preprint]. 2024 Nov 1:2024.10.29.620637. doi: 10.1101/2024.10.29.620637. bioRxiv. 2024. Update in: Mol Syst Biol. 2025 Oct;21(10):1325-1350. doi: 10.1038/s44320-025-00127-z. PMID: 39553989 Free PMC article. Updated. Preprint.
References
-
- Aguilera A, García-Muse T (2012) R loops: from transcription byproducts to threats to genome stability. Mol Cell 46:115–124 - PubMed
-
- Alsina A, Fumasoni M, Sartori P (2025) Model-based inference of cell cycle dynamics captures alterations of the DNA replication programme. bioRxiv10.1101/2025.03.19.644216v1
MeSH terms
Substances
Grants and funding
- UI/BD/152252/2021/MEC | Fundação para a Ciência e a Tecnologia (FCT)
- 2023.09068.CEECIND/MEC | Fundação para a Ciência e a Tecnologia (FCT)
- 2022.07846.PTDC/MEC | Fundação para a Ciência e a Tecnologia (FCT)
- 101030203/EC | Horizon Europe | Excellent Science | HORIZON EUROPE Marie Sklodowska-Curie Actions (MSCA)
- 5349-2023/European Molecular Biology Organization (EMBO)
LinkOut - more resources
Full Text Sources
