

In Silico Analysis of Phosphomannomutase-2 Dimer Interface Stability and Heterodimerization with Phosphomannomutase-1
- PMID: 40572562
- PMCID: PMC12195792
- DOI: 10.3390/molecules30122599
In Silico Analysis of Phosphomannomutase-2 Dimer Interface Stability and Heterodimerization with Phosphomannomutase-1
Abstract
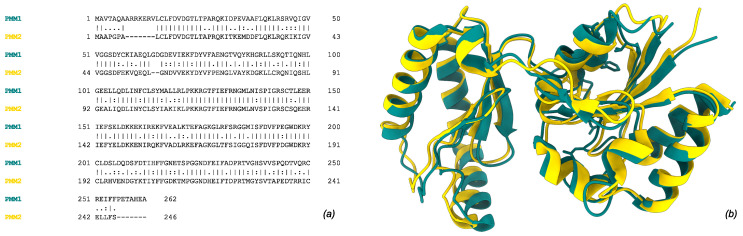
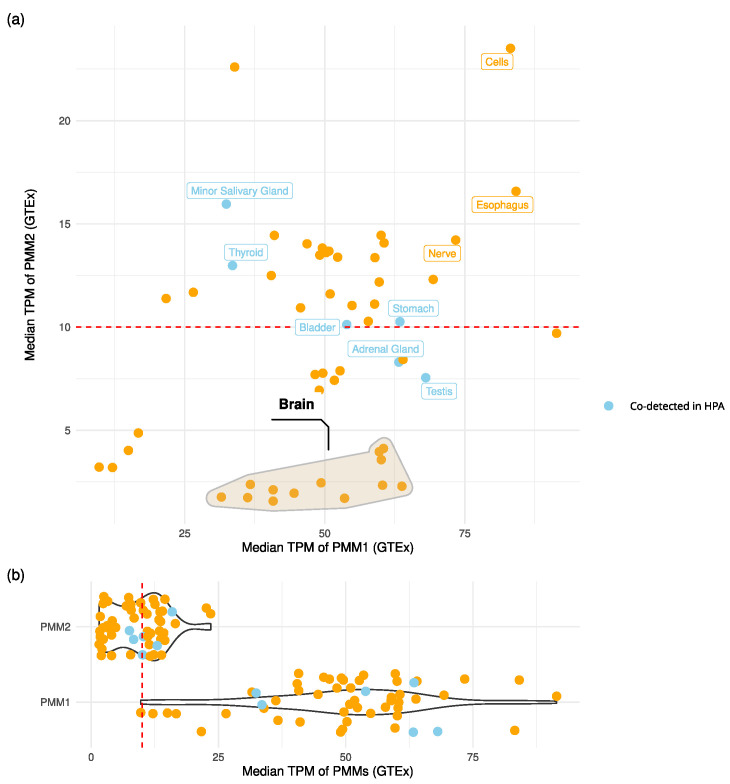
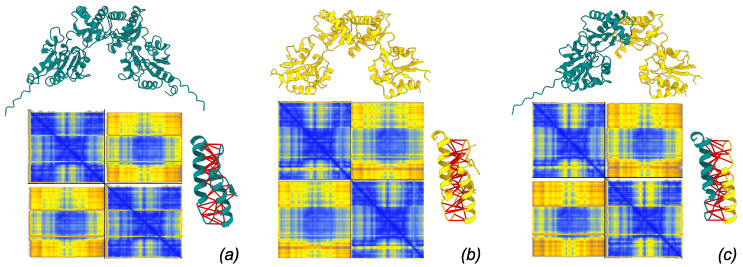
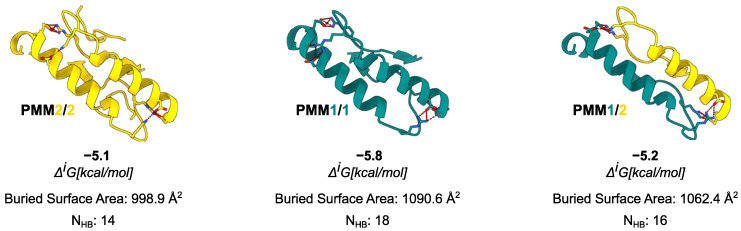
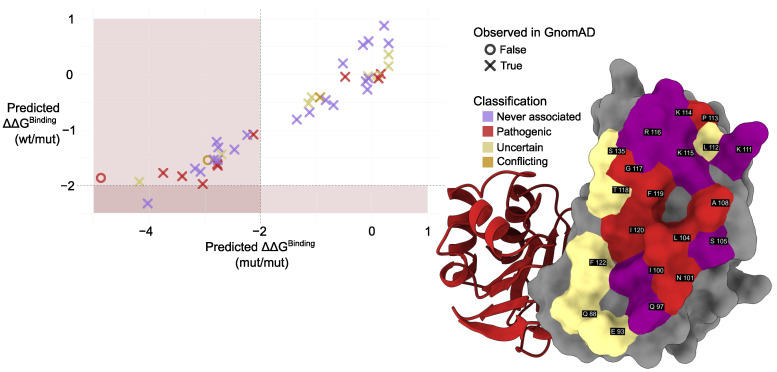
Phosphomannomutase 2 (PMM2) catalyzes the interconversion of mannose-6-phosphate and mannose-1-phosphate, a key step in the biosynthesis of GDP-mannose for N-glycosylation. Its deficiency is the most common cause of congenital disorders of glycosylation (CDGs), accounting for the subtype known as PMM2-CDG. PMM2-CDG is a rare autosomal recessive disease characterized by multisystemic dysfunction, including cerebellar atrophy, peripheral neuropathy, developmental delay, and coagulation abnormalities. The disease is associated with a spectrum of pathogenic missense mutations, particularly at residues involved in dimerization and catalytic function (i.e., p.Phe119Leu and p.Arg141His). The dimerization of PMM2 is considered essential for enzymatic activity, although it remains unclear whether this supports structural stability alone, or whether both subunits are catalytically active-a distinction that may affect how mutations in each monomer contribute to overall enzyme function and disease phenotype. PMM2 has a paralog, phosphomannomutase 1 (PMM1), which shares substantial structural similarity-including obligate dimerization-and displays mutase activity in vitro, but does not compensate for PMM2 deficiency in vivo. To investigate potential heterodimerization between PMM1 and PMM2 and the effect of interface mutations over PMM2 dimer stability, we first assessed the likelihood of their co-expression using data from GTEx and the Human Protein Atlas. Building on this expression evidence, we modeled all possible dimeric combinations between the two paralogs using AlphaFold3. Models of the PMM2 and PMM1 homodimers were used as internal controls and aligned closely with their respective reference biological assemblies (RMSD < 1 Å). In contrast, the PMM2/PMM1 heterodimer model, the primary result of interest, showed high overall confidence (pLDDT > 90), a low inter-chain predicted alignment error (PAE∼1 Å), and robust interface confidence scores (iPTM = 0.80). Then, we applied PISA, PRODIGY, and mmCSM-PPI to assess interface energetics and evaluate the impact of missense variants specifically at the dimerization interface. Structural modeling suggested that PMM2/PMM1 heterodimers were energetically viable, although slightly less stable than PMM2 homodimers. Interface mutations were predicted to reduce dimer stability, potentially contributing to the destabilizing effects of disease-associated variants. These findings offer a structural framework for understanding PMM2 dimerization, highlighting the role of interface stability, paralogs co-expression, and sensitivity to disease-associated mutations.
Keywords: PMM2-CDG; PPI; human PMMs; structural bioinformatics.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

References
-
- Sharma V., Freeze H. Phosphomannomutase 1,2 (PMM1,2) In: Taniguchi N., Honke K., Fukuda M., Narimatsu H., Yamaguchi Y., Angata T., editors. Handbook of Glycosyltransferases and Related Genes. Springer; Tokyo, Japan: 2014. pp. 1591–1598. - DOI
-
- Schollen E., Pardon E., Heykants L., Renard J., Doggett N.A., Callen D.F., Cassiman J.J., Matthijs G. Comparative Analysis of the Phosphomannomutase Genes PMM1, PMM2 and PMM2psi: The Sequence Variation in the Processed Pseudogene is a Reflection of the Mutations Found in the Functional Gene. Hum. Mol. Genet. 1998;7:157–164. doi: 10.1093/hmg/7.2.157. - DOI - PubMed
-
- Lam C., Krasnewich D.M. GeneReviews® [Internet] University of Washington; Seattle, WA, USA: 2021. PMM2-CDG.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
