

Synergistic Autophagy-Related Mechanisms of Protection Against Brain Aging and AD: Cellular Pathways and Therapeutic Strategies
- PMID: 40573225
- PMCID: PMC12195936
- DOI: 10.3390/ph18060829
Synergistic Autophagy-Related Mechanisms of Protection Against Brain Aging and AD: Cellular Pathways and Therapeutic Strategies
Abstract
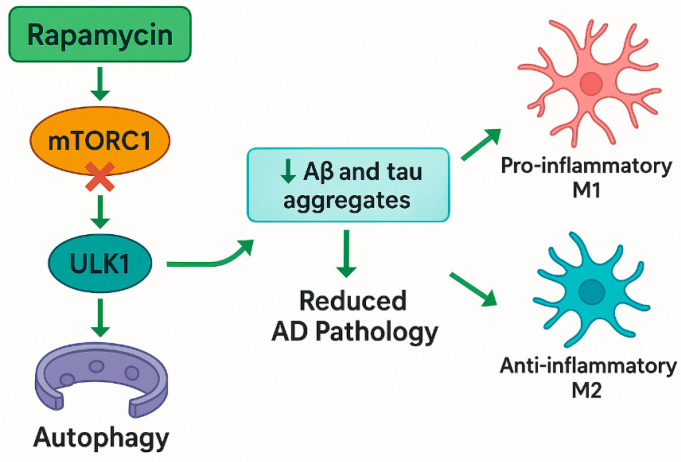
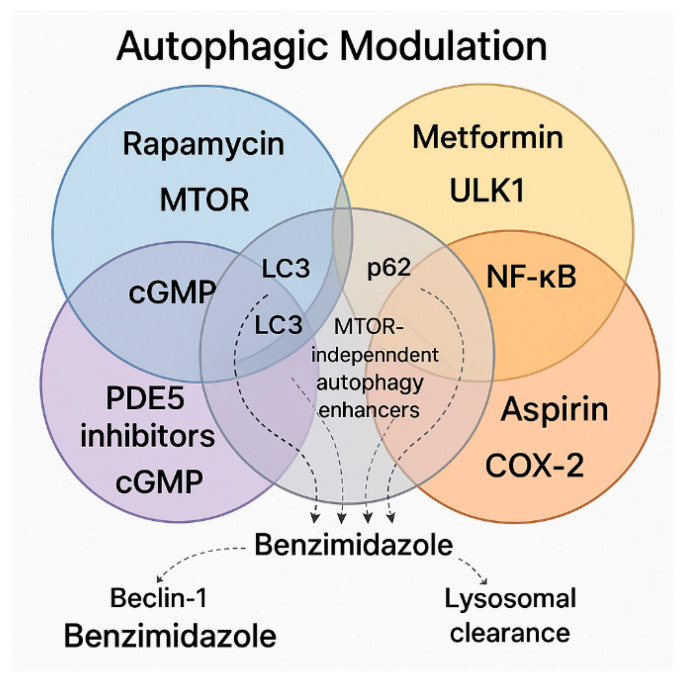
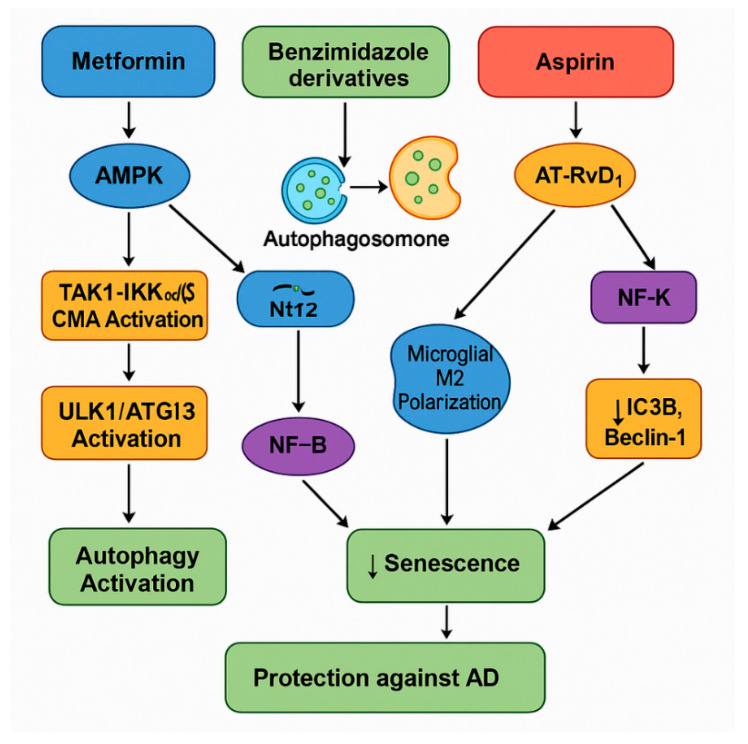
Brain aging is driven by interconnected processes, including impaired autophagy, chronic inflammation, mitochondrial dysfunction, and cellular senescence, all of which contribute to neurovascular decline and neurodegenerative diseases such as Alzheimer's disease (AD). Targeting these mechanisms simultaneously offers a promising therapeutic approach. This review explores the rationale for combining metformin, benzimidazole derivatives, phosphodiesterase-5 (PDE5), and acetylsalicylic acid (ASA) as a multi-targeted strategy to restore proteostasis, reduce senescence-associated secretory phenotype (SASP) factors, and enhance mitochondrial and lysosomal function. Metformin activates AMP-activated protein kinase (AMPK) and promotes autophagy initiation and chaperone-mediated autophagy, whilst benzimidazole derivatives enhance lysosomal fusion through JIP4-TRPML1 pathways independently of mTOR signaling; and ASA augments autophagic flux while suppressing NF-κB-driven inflammation and promoting specialized pro-resolving mediator pathways. This combinatorial approach targets both upstream autophagy initiation and downstream autophagosome-lysosome fusion, while concurrently attenuating inflammation and cellular senescence. Patient stratification based on the biomarkers of autophagy impairment, inflammation, and metabolic dysfunction could optimize therapeutic responses. While this strategy shows strong preclinical promise, careful attention to timing, dosing, and cell-specific responses is crucial to maximize benefits and avoid adverse effects. Future studies integrating biomarker-guided precision medicine frameworks are essential to validate the potential of this therapeutic combination in preventing or slowing cognitive decline and promoting healthy brain aging.
Keywords: Alzheimer’s disease; acetylsalicylic acid; autophagy; benzimidazole; metformin; neurodegeneration.
Conflict of interest statement
Authors Ian Hampson and Anthony Oliver were employed by the company Ravan Bio Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors declare that this study received no funding from either Ravan Bio Ltd., or the Caring Cancer Trust and The Cancer Prevention Research Trust, UK. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Figures




References
-
- Ting K.K., Coleman P., Kim H.J., Zhao Y., Mulangala J., Cheng N.C., Li W., Gunatilake D., Johnstone D.M., Loo L., et al. Vascular Senescence and Leak Are Features of the Early Breakdown of the Blood–Brain Barrier in Alzheimer’s Disease Models. GeroScience. 2023;45:3307–3331. doi: 10.1007/s11357-023-00927-x. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
